Lexikon der Chemie: Stereoisomerie
Stereoisomerie, Raumisomerie, besondere Art von Isomerie. Stereoisomere sind Isomere, die sich bei gleicher Konstitution nur in der Anordnung der Atome und Atomgruppen im Raum unterscheiden. Die Stereoisomerie wird im Rahmen der Stereochemie behandelt und folgendermaßen unterteilt:
;)
1. Konfigurationsisomerie. Konfigurationsisomere sind Stereoisomere, die sich in der räumlichen Anordnung von Atomen und Atomgruppen unterscheiden, ohne Berücksichtigung von Orientierungen, die nur durch Rotation um Einfachbindungen voneinander abweichen. Hierzu gehören Anordnungen an Doppelbindungen, Ringverbindungen und Chiralitätselementen. Die meisten Konfigurationsisomere können im Unterschied zu den Konformationsisomeren (s. u.) isoliert werden.
1.1. Enantiomerie. Enantiomere sind Konfigurationsisomere, die sich wie Bild und Spiegelbild verhalten. Sie zeigen unabhängig vom Aggregatzustand und auch in Lösung optische Aktivität, d. h., sie drehen die Ebene des linear-polarisierten Lichtes beim Durchgang durch sie, und zwar um den gleichen Betrag, aber im entgegengesetzten Sinn. Die rechtsdrehende Form wird durch ein dem Namen vorangestelltes (+) die linksdrehende durch ein (-) gekennzeichnet (Polarimetrie). Als Bezugssystem dient Glycerinaldehyd (Abb. 1).
;)
Stereoisomerie. Abb. 1: Die beiden Enantiomeren des Glycerinaldehyds
Außer in der Drehrichtung der Ebene des linear polarisierten Lichtes unterscheiden sich Enantiomere noch in der Kristallform, in der physiologischen Wirkung und in der Reaktion mit einem Enantiomeren einer anderen Verbindung. In allen anderen physikalischen Eigenschaften und in den chemischen Reaktionen mit nicht optisch aktiven Verbindungen stimmen sie überein. Reine Enantiomere wurden erstmals von Pasteur am Beispiel der Weinsäure gefunden.
Von einer chem. Verbindung existieren zwei Enantiomere, wenn sie mit ihrem Spiegelbild nicht zur Deckung gebracht werden kann. Diese Inkongruenz von Molekül und Spiegelbild liegt vor, wenn das betreffende Molekül weder eine Spiegelebene (σ) noch ein Inversionszentrum (i), noch eine Drehspiegelachse (Sn) aufweist. (Eine einfache Drehachse (Cn) kann vorhanden sein.) Solche Moleküle haben die topologische Eigenschaft der Händigkeit (Chiralität), die demzufolge ein notwendiges und hinreichendes Kriterium für die Existenz von Enantiomeren und damit für optische Aktivität ist. Über die Beziehungen zwischen Symmetrie, Chiralität und optischer Aktivität informiert die Tabelle. Die Chiralität einer Verbindung wird durch ein Chiralitätselement bewirkt. Die überwiegende Mehrzahl aller optisch aktiven, also chiralen Verbindungen enthält ein asymmetrischesKohlenstoffatom, d. i. ein Kohlenstoffatom mit vier verschiedenen Substituenten, als Chiralitätszentrum bzw. stereogenes Zentrum. Glycerinaldehyd, Milchsäure und Weinsäure sowie fast alle Naturstoffe, z. B. Kohlenhydrate, Eiweiße, Steroide und Alkaloide, enthalten asymmetrische Kohlenstoffatome (Abb. 2). Daneben können auch andere asymmetrisch substituierte Zentralatome Chiralitätszentren sein, z. B. Silicium in Silanen, Stickstoff in quartären Ammoniumsalzen und Aminoxiden, Schwefel in Sulfoxiden sowie Metallatome (M) in Komplexverbindungen (Abb. 3). Chiralitätsachsen enthalten entsprechend substituierte Allene, Spirane und Diphenylderivate (Abb. 4). Eine Chiralitätsebene weist trans-Cycloocten auf (Abb. 5).
;)
Stereoisomerie. Abb. 2: Asymmetrische Kohlenstoffatome (durch * gekennzeichnet) als Chiralitätszentren.
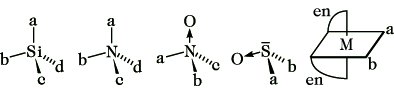
Stereoisomerie. Abb. 3: Si, N, S und M als Chiralitätszentren.
Die Helizität ist ein Sonderfall der Chiralität, bei der die Enantiomerie durch den Schraubensinn einer Achse mit Gang (Konfigurationsbezeichnung: M oder P) besonders bei Makromolekülen und angular kondensierten Aromaten (Helicene) charakterisiert wird (Abb. 6).
Stereoisomerie. Tab.: Beziehungen zwischen Symmetrie, Chiralität und optischer Aktivität.
| ||||
| Achiral, symmetrisch | + | + | – | |
| Chiral, axial symmetrisch | + | – | + | |
| Chiral, asymmetrisch | – | – | + |
;)
Stereoisomerie. Abb. 4 Moleküle mit Chiralitätsachse.
;)
Stereoisomerie. Abb. 5: Molekül mit Chiralitätsebene.
Sind an einem Zentralatom zwei verschiedene achirale Liganden und zwei konstitutionell gleiche, aber chirale Liganden gebunden, so spricht man von Pseudoasymmetrie.
Eine besondere Form ist die geometrische Enantiomerie, die auftritt, wenn an Kohlenstoffdoppelbindungen an dem einen C-Atom zwei verschiedene achirale Gruppen und am anderen zwei chirale entgegengesetzter Konfiguration ( ![]()
und ![]()
) gebunden sind (Abb. 7).
;)
Stereoisomerie. Abb. 6: Hexahelicen. (P)- Abk. von Plus, (M)- Abk. von Minus.
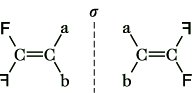
Stereoisomerie. Abb. 7: Geometrische Enantiomere.
Die beiden Enantiomeren einer chiralen Verbindung unterscheiden sich in der absoluten Konfiguration, die erstmals 1951 von Bijvoet durch Röntgenbeugung am Rubidiumsalz der Weinsäure bestimmt wurde. Zur Kennzeichnung der Enantiomeren benutzt man relativeKonfigurationsbezeichnungen. Die im Polarimeter gemessene Drehrichtung hängt nicht von der Konfiguration ab, z. B. dreht das Natriumsalz der rechtsdrehenden Milchsäure nach links, und ist deshalb zur Bezeichnung der Konfiguration nicht geeignet.
Bei der Konfigurationsbezeichnung nach E.
Fischer schreibt man die betrachteten Enantiomeren in der Fischer-Projektion (Stereochemie) so, daß die längste C-Kette senkrecht angeordnet ist und die höchste Oxidationsstufe oben steht, dann hat das Enantiomere, bei dem der waagerecht angeordnete Substituent rechts steht, die D-, das andere die L-Konfiguration (Abb. 1). Diese ältere Konfigurationsbezeichnung wird z. B. noch bei Naturstoffen verwendet, sie versagt, wenn nicht mehr zu entscheiden ist, was die längste C-Kette, die höchste Oxidationsstufe oder was Substituent ist (z. B. bei CFClBrI).
Die Konfigurationsbezeichnung nach der Cahn-Ingold-Prelog-Konvention (CIP-Priorität) (Abb. 8) ist eindeutig, logisch und für alle chiralen Verbindungen anwendbar. Man ordnet zunächst die Liganden eines Chiralitätszentrums nach fallenden Ordnungszahlen (Sequenz: a > b > c > d). Doppelbindungen werden zu zwei Einfachbindungen mit jeweils denselben Substituenten aufgelöst. Wenn in der ersten Sphäre keine Priorität feststellbar ist, muß sie in der zweiten oder einer höheren gefunden werden. Betrachtet man nun das Molekül von der Gegenseite des Substituenten d, dann hat das Enantiomere, bei dem die Reihenfolge a → b → c eine Rechtsdrehung (Uhrzeigersinn) ergibt , die R-, das andere die S-Konfiguration. In Verbindungen mit Pseudosymmetriezentren wird die Konfiguration mit r bzw. s gekennzeichnet.
;)
Stereoisomerie. Abb. 8: Konfiguration nach Cahn-Ingold-Prelog.
Äquimolekulare Gemische zweier Enantiomere einer Verbindung werden Racemate genannt. Die Zusammensetzung anderer Gemische gibt man durch den Enantiomerenüberschuß e.e. oder das Enantiomerenverhältnis an.
1.2. Diastereomerie. Diastereomere sind alle Konfigurationsisomeren, die nicht Enantiomere sind. Sie treten bei Verbindungen mit mehreren Chiralitätselementen auf, sie können bei alicyclischen Verbindungen, bei Verbindungen mit Doppelbindungen und an Komplexverbindungen auftreten.
1.2.1. Diastereomerie aufgrund mehrerer Chiralitätselemente. Enthält eine Verbindung n Chiralitätselemente (meist asymmetrische Kohlenstoffatome), so können maximal 2n Konfigurationsisomere existieren. Diejenigen, die sich in der Konfiguration aller asymmetrischen Kohlenstoffatome unterscheiden, sind Enantiomere (es gibt 2n/2 Enantiomerenpaare). Alle anderen, die mindestens in der Konfiguration eines asymmetrischen Kohlenstoffatoms gleich sind und sich mindestens in der Konfiguration an einem asymmetrischen Kohlenstoffatom unterscheiden, sind Diastereomere. Verbindungen mit zwei strukturell nicht gleichen asymmetrischen Kohlenstoffatomen (z. B. die Aldotetrosen) existieren demzufolge in 4 Konfigurationsisomeren. Dabei sind jeweils (2R,3R)- und (2S,3S)-Erythrose sowie (2R,3S)- und (2S,3R)-Threose Enantiomere, alle 4 anderen möglichen Beziehungen sind die von Diastereomeren (Abb. 9).
;)
Stereoisomerie. Abb. 9: Enantiomerie und Diastereomerie am Beispiel der Aldotetrosen.
Zur Kennzeichnung von Diastereomerenpaaren mit benachbarten Chiralitätszentren benutzt man die Vorsilben erythro, wenn in der Fischer-Projektion beide Substituenten in die gleiche Richtung zeigen, z. B. in der Erythrose (in der Newman-Projektion lassen sich die jeweils gleichen Substituenten durch entsprechende Drehung gleichzeitig zur Deckung bringen), und threo, wenn die Substituenten nach verschiedenen Seiten zeigen, z. B. in der Threose (in der Newman-Projektion lassen sich die beiden gleichen Substituenten nicht gleichzeitig zur Deckung bringen). Die Zahl von 2n Konfigurationsisomeren verringert sich, wenn strukturgleiche Chiralitätszentren, d. h. asymmetrische Kohlenstoffatome mit je 4 gleichen Substituenten, vorliegen, z. B. in der Weinsäure (Abb. 10). (2R,3R)- und (2S,3S)-Weinsäure sind Enantiomere, die dazu diastereomere (2R,3S)-Weinsäure ist optisch inaktiv. Letztere hat eine Symmetrieebene, die Drehungen der beiden Molekülhälften kompensieren sich innerhalb des Moleküls. Man bezeichnet solche Verbindungen als innereRacemate (sie lassen sich nicht in Enantiomere spalten) und kennzeichnet sie durch die Vorsilbe meso. Liegen Diastereomere mit 2 Chiralitätszentren vor, die nur einen oder keinen gleichen Substituenten tragen, bezeichnet man die beiden Diastereomeren mit L- (like, wenn beide Chiralitätszentren die (R)- oder (S)-Konfiguration haben) und mit u- (unlike, wenn ein Chiralitätszentrum die (R)-, das andere die (S)- Konfiguration besitzt).
;)
Stereoisomerie. Abb. 10: Weinsäure.
Epimere sind Diastereomere bei Verbindungen mit mehreren asymmetrischen Kohlenstoffatomen, die sich nur in der Konfiguration eines dieser asymmetrischen Kohlenstoffatome unterscheiden (z. B. sind D-Glucose und D-Mannose Epimere). Unter Epimerisierungversteht man demzufolge die Konfigurationsumkehr nur eines von mehreren asymmetrischen Kohlenstoffatomen. Handelt es sich dabei um die Konfiguration am C1-Atom der Monosaccharide, die glycosidische OH-Gruppe in der Cyclohalbacetalform betreffend, spricht man von Anomeren, die man als α- und β-Form (Mutarotation) bezeichnet.
Diastereomere haben unterschiedliche chemische und physikalische Eigenschaften, sie lassen sich demzufolge durch Destillation, Kristallisation oder andere Trennverfahren voneinander trennen. Davon macht man bei der Racemattrennung (Racemat) durch intermediäre Bildung von Diastereomeren aus einem Racemat und einer optisch aktiven Verbindung Gebrauch.
Die Zusammensetzung von Diastereomerengemischen gibt man durch den Diastereomerenüberschuß d.e. oder das Diastereomerenverhältnis an.
1.2.2. Diastereomerie bei cyclischen Verbindungen tritt auf, wenn diese mindestens zwei Substituenten enthalten, z. B. Dichlorocyclopentane. Diese Diastereomere können mit R und S, jedoch üblicherweise durch die Präfixe cis (Abk. c; beide Substituenten auf der gleichen Seite des Cyclus) und trans (Abk. t; beide Substituenten auf verschiedenen Seiten) gekennzeichnet werden. Bei mehr als zwei Substituenten muß man einen Referenzsubstituenten (r) angeben (Abb. 11). Die Präfixe cis und trans sind auch zur Kennzeichnung der Verknüpfung von zwei Cyclen möglich, z. B. cis- und trans-Decalin). Bei Bicyclen verwendet man die Bezeichnungen endo und exo, je nachdem, ob der Substituent cis oder trans zur am höchsten bezifferten Brücke angeordnet ist (Abb. 12).
;)
Stereoisomerie. Abb. 11: Diastereomerie bei substituierten Cyclopentanen.
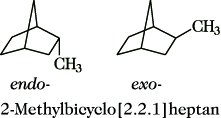
Stereoisomerie. Abb. 12: Diastereomerie bei Bicyclen.
1.2.3. Diastereomerie bei Verbindungenmit Doppelbindungen wird als cis-trans-Isomerie oder E,Z-Isomerie bezeichnet. Sie tritt auf, wenn an beiden Enden der Doppelbindung je zwei verschiedene Substituenten gebunden sind: (a,b)C=C(c,d). Für den Fall (a,b)C=C(a,b) oder (a,b)C=C(a,c) unterscheidet man je nach Stellung der beiden Substituenten a zueinander in bezug auf die Doppelbindungsebene zwischen cis- und trans-Form. lm allgemeinen Fall (a,b)C=C(c,d) (auch für die oben genannten Spezialfälle anwendbar) verwendet man zur Bezeichnung der zwei Diastereomeren die Sequenzregel nach der Cahn-Ingold-Prelog-Konvention und bezeichnet dasjenige mit den Substituenten gleicher Priorität auf derselben Seite als Z-Form (Abk. von zusammen) und dasjenige mit den Substituenten gleicher Priorität auf entgegengesetzter Seite als E-Form (Abk. von entgegen). Diese Kennzeichnungen gelten ebenso für Doppelbindungen an Stickstoffatomen, wo auch die Bezeichnungen syn- und anti- verwendet werden (Abb. 13). Die Bezeichnungen cis, syn und Z bzw. trans, anti und E sind nicht immer synonym.
;)
Stereoisomerie. Abb. 13: Diastereomerie bei Verbindungen mit Doppelbindung.
Die Konfiguration ermittelt man aus physikalischen Daten (z. B. hat meistens die cis-Form den niedrigeren Schmelzpunkt, die höhere Löslichkeit und das größere Dipolmoment), spektroskopischen Messungen, besonders aus IR-, UV- und NMR-Spektren sowie chemischen Reaktionen (z. B. gibt die Maleinsäure ein inneres Anhydrid, die Fumarsäure nicht). Im Normalfall ist die trans-Form die thermodynamisch stabilere, bedingt durch elektronische und sterische Wechselwirkungen. Ausnahmen bilden z. B. cis-1,2-Dichlorethen und cis-Cycloocten. Die cis,trans-Isomerisierung ist durch thermische und photochemische Energiezufuhr (Entkopplung der π-Bindung) oder katalytisch (intermediäre Addition von Radikalen oder Ionen) möglich. cis,trans-Isomerie tritt auch bei Koordinationsverbindungen auf.
2. Konformationsisomerie.Konformationsisomere (Konformere, Rotationsisomere, Rotamere) sind Stereoisomere, die sich bei gleicher Konfiguration nur durch Rotation um C-C-Einfachbindungen voneinander unterscheiden. Sie können nur isoliert werden, wenn die Rotation um die betrachtete formale Einfachbindung verhindert ist (Atropisomerie).
2.1. Konformation acyclischer Verbindungen.Bereits die Rotation um die C-C-Bindung im Ethan ist nicht völlig frei möglich. Bei der Drehung um 360° treten drei Energiebarrieren (Rotationsbarrieren) von 11,7 kJ/mol auf, die den Energieunterschied zwischen zwei ausgezeichneten Konformationen darstellen, der gestaffelten und der ekliptischen (verdeckten) Konformation (Abb. 14). Energieärmer ist die gestaffelte Form, Ursache des höheren Energieinhaltes der ekliptischen Konformation ist die Abstoßung der H-Atome einschließlich der Elektronenwolken (Pitzer-Spannung, Torsionsspannung). Bei Rotation um die mittlere C-C-Bindung des n-Butans treten verschiedene ausgewählte Konformationen auf, deren Energieunterschiede und Bezeichnungen aus Abb. 15 hervorgehen.
;)
Stereoisomerie. Abb. 14: Konformation des Ethans: (a) in der perspektivischen Stereoformel, (b) in der Newman-Projektion.
;)
Stereoisomerie. Abb. 15: Konformationen des n-Butans.
Aus den Konformationsbetrachtungen acyclischer Verbindungen ergeben sich folgende Schlußfolgerungen: a) Die Drehbarkeit um C-C-Einfachbindungen ist vorhanden, sie ist jedoch nicht völlig frei, es treten Rotationsbarrieren auf. b) Im Konformationsgleichgewicht sind die Konformationen bevorzugt, die die geringste Konformationsenergie (Summe aller Energiewerte, die die Stabilität von Konformationen beeinflussen) haben. Die größten Substituenten nehmen die antiperiplanare Konformation ein, daraus folgt für n-Alkane die Zickzackanordnung als Vorzugskonformation. c) Durch entgegengesetzt geladene Gruppen oder durch Wasserstoffbrücken kann die gauche-Konformation bevorzugt werden. d) Nur in einigen seltenen Fällen, z. B. bei 2,2',6,6'-tetra-substituierten Diphenylderivaten (Atropisomerie) und bei cyclisch miteinander verknüpften Gruppen (Ansaverbindungen) sind die Rotationsbarrieren so hoch, daß unter normalen Bedingungen eine Rotation nicht möglich ist.
Die Untersuchung solcher Konformationsgleichgewichte und die Abschätzung von Stabilitätsverhältnissen einzelner Konformationsisomerer mittels physikalischer Daten (besonders Dipolmoment) und spektroskopischer Methoden (besonders NMR-Spektroskopie) bezeichnet man als Konformationsanalyse.
2.2. Konformation des Cyclohexans.Besondere Bedeutung hat die Konformationsanalyse für cyclische Verbindungen. Im Gegensatz zur ursprünglichen Annahme von Baeyer (1885, Baeyersche Spannungstheorie) sind die stabilen Konformationen der Cycloalkane nicht planar. Schon 1890 schlug Sachse den nichtebenen Bau des Cyclohexans und anderer Cycloalkane vor. Nach der Theorie von Sachse und Mohr existiert das Cyclohexan in zwei nicht planaren bindungswinkelspannungsfreien Konformationen, der Sessel- und der Boot- oder Wannenkonformation (Abb. 16). Die Sesselform hat eine dreizählige Drehachse und eine sechszählige Drehspiegelachse, sie ist starr und 23 kJ/mol stabiler als die Bootform, es treten nur gauche-Konformationen auf. Die Bootform (Wannenform) ist energiereicher als die Sesselform, neben vier gauche-Konformationen gibt es zwei ekliptische, synperiplanare, und darüber hinaus treten Abstoßungskräfte zwischen den 1,4-ständigen H-Atomen auf. Die Bootform ist flexibel, durch Pseudorotation geht sie in alternative Bootformen über, dabei wird eine um etwa 3 kJ/mol stabilere Konformation, die Twist-Bootform (Twistform) durchlaufen (Abb. 16). Aufgrund des Energieunterschiedes zwischen Sessel- und Bootform liegt das Cyclohexan bei Raumtemperatur zu 99,9% in der Sesselform vor. Die Bootform kann in Bicyclen oder durch Anziehungskräfte zwischen den 1,4-ständigen Substituenten (z. B. H-Brücken) stabilisiert werden (Abb. 17).
;)
Stereoisomerie. Abb. 16: Konformationen des Cyclohexans.
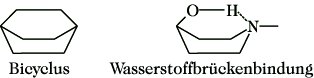
Stereoisomerie. Abb. 17: Stabilisierung der Boot-Konformation.
Der Übergang der Sesselform in die Twist- oder Bootform oder aber auch in eine Alternativ-Sesselform ist durch Ringinversion möglich (Abb. 18). Die Inversionsbarriere, die einer Halbsesselform entspricht, beträgt 46 kJ/mol. An jedem Kohlenstoffatom des Cyclohexans sind noch zwei Bindungen frei. In der Sesselform sind es 6 axiale Bindungen (Symbol a), die parallel zur Symmetrieachse abwechselnd nach oben und unten gerichtet sind, und 6 äquatoriale Bindungen (Symbol e), die mit der Achse einen Winkel von 70° bilden und ebenfalls abwechselnd nach oben und unten zeigen (Abb. 16). Axial gebundene Substituenten stehen zum Cyclohexangerüst in gauche-Konformation, äquatorial gebundene in anti-periplanarer. Axiale Substituenten unterliegen darüber hinaus einer 1,3-Wechselwirkung. Die äquatoriale Anordnung ist demzufolge die thermodynamisch stabilere. Axiale und äquatoriale Bindungen sind durch Ringinversion ineinander umwandelbar. Große Gruppen bevorzugen deshalb die äquatoriale Anordnung. Der Raumbedarf z. B. der tert-Butylgruppe ist so groß, daß das tert-Butylcyclohexan weitestgehend in der äquatorialen Konformation vorliegt. Eine solche Gruppe, die bestimmte Konformationen fixiert, wird als Konformationsanker bezeichnet.
;)
Stereoisomerie. Abb. 18: Ringinversionen des Cyclohexans (a) und des substituierten Cyclohexans (b).
2.3. Konformation anderer Ringsysteme (Abb. 19). Das Cyclobutan ist gewinkelt, dadurch wird die ekliptische Stellung der H-Atome beseitigt, Alternativkonformationen entstehen durch Pseudorotation. Der Energiegehalt wird wie im Cyclopropan im wesentlichen durch Bindungswinkelspannung bedingt. Cyclopentan und Cycloheptan liegen in solchen Konformationen vor, z. B. Halbsesselform bzw. verdrillte Sesselform, die den energetischen Kompromiß zwischen Bindungswinkel- und Torsionsspannung darstellen. Pseudorotationen bewirken auch in diesen beiden Fällen die schnelle gegenseitige Umwandlung von Alternativkonformationen.
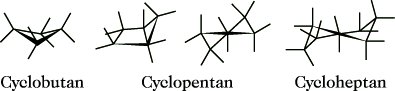
Stereoisomerie. Abb. 19: Konformationen verschiedener Ringsysteme.
Bei mittleren Ringen (C8 bis C11) existieren jeweils mehrere Konformationsisomere ähnlicher Konformationsenergie, die jeweils der Kompromiß zwischen Bindungswinkel- und Torsionsspannung sowie einer zusätzlichen Spannung (transannulare Spannung) sind. Solche transannularen Spannungen sind das Ergebnis von nichtbindenden Wechselwirkungen im Inneren der mittleren Ringe durch Pressungen der Van-der-Waals-Radien innerer H-Atome. Die großen Ringe (>C11) liegen als Doppelketten in Zickzackkonformation vor.








Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.