Lexikon der Chemie: UV-VIS-Spektroskopie
UV-VIS-Spektroskopie (Abk. von engl. ultraviolet and visible spectroscopy), UVS-Spektroskopie, Teilgebiet der Spektroskopie, das auf einer Wechselwirkung elektromagnetischer Strahlung aus dem ultravioletten und sichtbaren Bereich mit einer Probe beruht. Da in beiden Bereichen die gleichen Anregungsprozesse, nämlich Elektronenübergänge, vor sich gehen, erfolgt eine Zusammenfassung der Ultraviolettspektroskopie (UV-Spektroskopie) und der Spektroskopie im Sichtbaren als UV-VIS-S.
Während das Gebiet des sichtbaren Lichtes (400 bis 800 nm) durch die Empfindlichkeit des menschlichen Auges festgelegt ist, wird das UV-Gebiet aus vorwiegend apparativen Gründen in das kürzerwellige Vakuum-UV oder ferne UV (10 bis 200 nm) und das längerwellige nahe UV oder Quarz-UV (200 bis 400 nm) unterteilt. Da unterhalb 200 nm die Luft UV-Strahlung absorbiert, sind Messungen in diesem Bereich nur in besonderen, evakuierbaren Spektro-
metern möglich. Die Bezeichnung Quarz-UV weist darauf hin, daß die optischen Bauelemente (Spektralapparaturen) im nahen UV meist aus Quarz bestehen.
Die Spektren von Atomen im UV-VIS werden sowohl als Absorptionsspektrcn (Atomabsorptionsspektrometrie) als auch als Emissionsspektren (Atomemissionsspektrometrie), die von Molekülen fast ausschließlich als Absorptionsspektren vermessen.
Absorptionsspektren von Molekülen:Molekülspektren können in allen 3 Aggregatzuständen vermessen werden. Es überwiegt die Aufnahme in absorptionsfreien Lösungsmitteln, wie Wasser, Alkohol, Alkanen. Die graphische Darstellung der Spektren erfolgt, indem man die Wellenlänge in nm – seltener die Wellenzahl in cm-1 – als Funktion der Intensität (% Absorption, % Durchlässigkeit, Extinktion ε oder molarer Extinktionskoeffizient) aufträgt. Der Substanzbedarf ist äußerst gering. Es werden Lösungen einer Konzentration von etwa 10-2 bis 10-5 mol l-1 benötigt.
Beobachtet man im Vergleich zu einer Referenzsubstanz bei den Absorptionsbanden im UV-VIS-Spektrum eine Erhöhung der Intensität, so spricht man von hyperchromer Verschiebung, bei Abnahme der Intensität von hypochromer Verschiebung. Verschiebt sich das Absorptionsmaximum nach längeren Wellenlängen, so liegt bathochrome Verschiebung vor, verschiebt es sich nach kürzeren Wellenlängen, bezeichnet man dies als hypsochrome Verschiebung.
Theorie. Schon lange, ehe die Kenntnisse über den Atombau eine physikalische Erklärung der Vorgänge bei der Lichtabsorption zuließen, hatten die Chemiker durch Auswertung eines umfangreichen experimentellen Materials gewisse empirische Beziehungen zwischen chem. Struktur und Lichtabsorption im UV-VIS gefunden (Farbstofftheorie). Atomgruppen mit Mehrfachbindungen (chromophore Gruppen) wurden als unbedingt notwendig für eine langwellige Lichtabsorption erkannt. Diese chromophoren Gruppen (mit Ausnahme der Azogruppe) absorbieren für sich allein im UV. Um ihre Absorption ins Sichtbare zu verschieben, müssen mehrere chromophore Gruppen zueinander konjugiert angeordnet oder mit auxochromen Gruppen verknüpft sein.
Quantentheorie der Lichtabsorption: In der UV-VISS. finden Elektronenübergänge zwischen Orbitalen verschiedener Energie statt, die gemäß der Beziehung hν = E2- E1 zur Absorption bestimmter Frequenzen führen. Diese Orbitale und ihre Energie können mit Hilfe der Quantenmechanik berechnet werden. Allerdings lassen sich die molekularen Eigenfunktionen und ihre Energiewerte für mehratomige Moleküle nur näherungsweise angeben. Die wichtigsten dafür angewandten Verfahren sind das Molekularorbital-, das Valenzstruktur- und das Elektronengasverfahren. Am häufigsten wird gegenwärtig das LCAO-MO-Verfahren angewandt, das Molekülorbitale als Linearkombination von Atomorbitalen beschreibt. So resultieren aus der Kombination der 1s Orbitale von zwei Wasserstoffatomen zwei Molekülorbitale des Wasserstoffs, von denen eins einem bindenden (Symbol σs), das andere einem antibindenden Orbital (Symbol σ*s) entspricht (Abb. 1). Bei der Linearkombination von 2 pz Atomorbitalen entstehen Molekülorbitale, die mit πp und π*p bezeichnet werden, wobei durch den hochgestellten Stern ein antibindendes Orbital bezeichnet wird. Die eingeführten Symbole werden meist zu σ, π,σ*, π* abgekürzt. Weiterhin gibt es nichtbindende Orbitale (n-Orbitale), auf denen sich die freien Elektronenpaare der Heteroatome befinden. Die relative Lage der Orbitalenergien zeigt Abb. 2. Die Energie eines nichtbindenden Orbitals ist im allg. größer als von π- und σ-Orbitalen, aber kleiner als die der antibindenden.
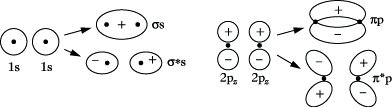
UV-VIS-Spektroskopie. Abb.1: Bindende und antibindende Molekülorbitale und ihre Bildung aus Atomorbitalen.
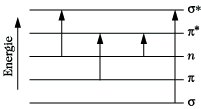
UV-VIS-Spektroskopie. Abb.2: Energien der Molekülorbitale und mögliche Übergänge zwischen ihnen.
Die möglichen Elektronenübergänge sind durch Pfeile eingezeichnet. Man erkennt, daß σ → π*, π → π* und π → σ* Übergänge weniger Energie erfordern als σ → σ* Übergänge. Letztere sind deshalb nur im Vakuum-UV zu beobachten, während die übrigen Übergänge im nahen UV und gegebenenfalls sogar im Sichtbaren auftreten können. Die relative Energie dieser Molekülorbitale kann durch verschiedene Faktoren beeinflußt werden. Die deutlichste Beeinflussung tritt bei Konjugation von Doppelbindtingen auf. Eine isolierte Doppelbindung gibt zu einem π → π>* Übergang bei etwa 180 nm Anlaß. In Dienen werden durch die Kombination der π-Orbitale der getrennten Alkene neue Orbitale gebildet, zwei bindende Orbitale π1 und π2 sowie zwei antibindende π3 und π4. Aus Abb. 3 ist ersichtlich, daß ein neuer π → π*-Übergang mit wesentlich geringerer Energie auftritt. Diene zeigen deshalb bei größeren Wellenlängen als Alkene eine Absorption. Die langweilige Verschiebung der Lichtabsorption setzt sich bei weiterer Konjugation von Doppelbindungen fort, wobei gleichzeitig die Intensität der Absorptionsbanden zunimmt (Tab. 1). Als Faustregel innerhalb einer Substanzklasse gilt: Je länger das konjugierte System, um so längerwellig liegt das Absorptionsmaximum.
;)
UV-VIS-Spektroskopie. Abb. 3: Änderung der Energie der Molekülorbitale des Ethens bei Konjugation.
UV-VIS-Spektroskopie. Tab. 1: Langwelligstes Absorptionsmaximum einiger Polyenaldehyde CH3-(CH=CH)n-CHO.
| |||
| 1 | 220 | 15000 | |
| 2 | 270 | 27000 | |
| 3 | 312 | 40000 | |
| 4 | 343 | 45000 | |
| 5 | 370 | 57000 | |
| 6 | 393 | 65000 | |
| 7 | 415 | 63000 |
Die Anregung von Elektronen wird von Schwingungs- und Rotationsübergängen begleitet. Jeder Elektronenübergang besteht aus mehreren Schwingungsteilbanden mit Rotationsfeinstruktur. Meist ist infolge zwischenmolekularer Wechselwirkungen diese Feinstruktur verwischt, und es ist lediglich ein breites Absorptionsmaximum erkennbar. In manchen Fällen, z. B. in der Gasphase und in unpolaren Lösungsmitteln, tritt die Schwingungsstruktur zuweilen auf. Man kann daraus auf Bindungskräfte und Bindungslängen der Moleküle im angeregten Zustand schließen.
Als Auswahlregeln für Elektronenübergänge gelten das Symmetrieverbot und das Spinverbot. Symmetrieverbot: Übergänge sind nur zwischen Elektronenzuständen bestimmter Symmetrie möglich. Aus der Symmetrie der Wellenfunktionen der miteinander kombinierenden Zustände kann abgeleitet werden, ob ein Elektronenübergang zwischen ihnen erlaubt oder verboten ist. Spinverbot: Übergänge zwischen Zuständen unterschiedlicher Multiplizität sind verboten.
Eine Desaktivierung angeregter Elektronenzustände ist möglich 1) durch Reemission als Strahlung, z. B. Fluoreszenz, Phosphoreszenz, 2) durch photochem. Reaktionen und 3) durch Umwandlung der Anregungsenergie in Wärmeenergie.
Anwendung. 1) Qualitative UV-VIS-S. Absorptionorganischer Substanzen. Vornehmlich werden konjugierte π-Elektronensysteme untersucht. So kann z. B. die Anzahl n der konjugierten Doppelbindungen in Polyenen R1-(C=C-)nR2 ermittelt werden. Für das längstwellige Absorptionsmaximum gilt die Beziehung
wobei A und B Konstanten sind, die von den Substituenten R1 und R2 abhängen.
Gesättigte Ketone und α,β-ungesättigte Ketone lassen sich anhand ihrer UV-Absorption unterscheiden. Während erstere ihre längstwellige Absorption bei etwa 270 nm aufweisen, ist dieser Übergang bei Konjugation mit einer Doppelbindung nach 320 nm verschoben. Mit Hilfe empirischer Regeln (Woodwardsche Absorptionsregeln) kann die Lichtabsorption für substituierte Diene und Triene sowie α,β-ungesättigte Ketone vorherberechnet werden.
Charakteristische Absorptionen im UV geben Benzol (Abb. 4) und seine Derivate. Wie Tab. 2 zeigt,
;)
UV-VIS-Spektroskopie. Abb. 4: Absorptionsspektrum des Benzols.
wird das Spektrum des Benzols durch Substituenten in charakteristischer Weise nach längeren Wellen verschoben. Auch das π-Elektronensystem der kondensierten Aromaten ergibt charakteristische Absorptionen, die sich hervorragend zur Identifizierung eignen (Abb. 5).
;)
UV-VIS-Spektroskopie. Abb. 5: Spektren kondensierter Aromaten: (a) Naphthalin, (b) Anthracen, (c) Naphthacen.
UV-VIS-Spektroskopie. Tab. 2: Absorptionsmaxima einiger monosubstituierter Benzole.
| ||||||
| H | 203 | 7400 | 254 | 204 | ||
| NH3+ | 203 | 7500 | 254 | 160 | ||
| CH3 | 206 | 7000 | 261 | 225 | ||
| Cl | 209 | 7400 | 263 | 190 | ||
| Br | 210 | 7900 | 261 | 192 | ||
| OH | 210 | 6200 | 270 | 1450 | ||
| COOH | 230 | 11600 | 273 | 970 | ||
| NH2 | 230 | 8600 | 280 | 1430 |
Eine ähnliche Abhängigkeit von der Anzahl der konjugierten Doppelbindungen wie in den Polyenen zeigt das längstwellige Absorptionsmaximum in Polymethinfarbstoffen, für die die Beziehung λmax= A n+ B gilt. Im Gegensatz zu den Polyenen besteht ein linearer Zusammenhang zwischen λ und n. Jede neue hinzukommende Doppelbindung verschiebt das Absorptionsmaximum um etwa 100 nm nach längeren Wellen. Sterische Hinderungen, die in konjugierten Systemen auftreten, können ebenfalls mit Hilfe der UV-VIS-S. erkannt werden. So wird das Absorptionsspektrum des Biphenyls durch Einführung von 2-Methyl-Substituenten nach kürzeren Wellenlängen verschoben. Ursache dafür ist die sterische Hinderung zwischen den Methylgruppen, die zu einer Verdrillung der beiden Benzolkerne und damit zu einer verminderten Konjugation zwischen ihnen führt.
Absorption anorganischer Verbindungen. Intensiv farbige Verbindungen kommen vor allem bei den Ionen der Übergangsmetalle und ihren Komplexen vor. Die Farbe beruht auf Übergängen der D-Elektronen bzw. der f-Elektronen in der Gruppe der Lanthanoide und Actinoide. Die D-D-Übergänge können mit Hilfe der Ligandenfeldtheorie verstanden werden. Neben den meist mit geringer Intensität vorkommenden D-D-Übergängen treten in Komplexen häufig charge-transfer-Übergänge auf, die eine wesentlich höhere Intensität aufweisen und meist im UV zu finden sind. Hier erfolgt der Übergang eines Elektrons von einem oxidierbaren Liganden zu einem Zentralatom in einem hohen Oxidationszustand. Die für diesen Elektronenübergang erforderliche Energie ist um so kleiner und die Absorption liegt bei um so größeren Wellenlängen, je stärker das Kation als Oxidationsmittel wirkt und je leichter oxidierbar der Ligand ist. Als Charakteristikum zeigen deshalb Übergangsmetallkomplexe intensive Absorptionen im UV (charge-transfer-Übergänge) und wesentlich schwächere Absorptionen im Sichtbaren (D-D-Übergänge) (Abb. 6). Absorptionen im UV können auch durch Elektronenübergänge in umgekehrter Richtung (von Liganden zum Zentralatom) auftreten, sowie durch π→π* oder n→π*-Übergänge innerhalb der Liganden hervorgerufen werden.
;)
UV-VIS-Spektroskopie. Abb. 6: Absorptionsspektrum von Ti(H2O)63+.
Verschiedene Elemente des Periodensystems, z. B. die Halogene, bilden farbige Moleküle. Die Farbigkeit beruht auf der Anregung von freien Elektronenpaaren. Auch Moleküle, die sich aus verwandten Elementen dieser Art zusammensetzen (z. B. S2Cl2), können farbig sein. Bestimmte Elemente, wie Kohlenstoff, Silicium, Bor, sind dunkel gefärbt. Diese Elemente sind Halbleiter, und ihre dunkle Farbe hängt damit zusammen.
2) Quantitative UV-VIS-S.Grundlage für quantitative Analysen ist das Lambert-Beersche Gesetz. Nach c= E/(εd) kann bei bekanntem molaren Extinktionskoeffizienten ε und bekannter Schichtdicke d die Konzentration c aus der gemessenen Extinktion E bestimmt werden. Auch Mehrkomponentenanalysen lassen sich mit Hilfe dieses Gesetzes durchführen. Mittels der quantitativen UV-VIS-S. können vor allem konzentrations- und temperaturabhängige Gleichgewichte (Tautomerie, Molekülassoziation, Komplexbildung, Thermochromie, Photochromie, Solvatochromie, Dissoziationskonstanten von Säuren und Basen) untersucht werden. Voraussetzung dafür ist, daß die im Gleichgewicht befindlichen Spezies ein unterschiedliches π-Elektronensystem aufweisen und sich somit in ihrer Absorption unterscheiden. So sind z. B. die Absorptionsspektren der beiden tautomeren Verbindungen
4-Nitrosophenol (mit benzoidem Charakter) und 1,4-Benzochinon-monoxim (mit chinoidem Charakter) deutlich unterschiedlich. Die quantitative Bestimmung zeigt, daß z. B. in etherischer Lösung ein Gleichgewicht von 70 % 1,4-Benzochinon-monoxim und 30 % 4-Nitroso-phenol vorliegt.
Für quantitative Bestimmungen im Rahmen der Photometrie ist die UV-VIS-S. ebenfalls von großer Bedeutung.
Zeitaufgelöste UV-VIS-S.Mit Routinespektrometern ist die Aufnahme eines UV-VIS-Spektrums in einigen Minuten möglich. Für die Untersuchung kurzlebiger Anregungs- und Zwischenprodukte sowie der Kinetik physikalischer, chem. und biochem. Prozesse sind jedoch Methoden mit einer wesentlich größeren Zeitauflösung erforderlich. In den 60er Jahren wurden Rapidspektroimeter entwickelt, die eine Zeitauflösung von 10-2 bis 10-5 s gestatteten. Durch die Blitzlichtspektroskopie wurde die Aufnahme von Spektren in 10-5 bis 10-7 s möglich. Durch Einsatz der Laser-Technik wurde die Nanosekunden- und Pikosekundenspektroskopie zugänglich. Mit Hilfe dieser Methoden hat die UV-VIS-S. in starkem Maße zur Entwicklung der Photophysik, Photochemie und der chem. Reaktionskinetik beigetragen.








Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.