Lexikon der Chemie: Alkene
Alkene, Olefine, Ethylenkohlenwasserstoffe, ungesättigte, C=C-Doppelbindungen enthaltende Kohlenwasserstoffe. Im weiteren Sinne sind A. Verbindungen mit einer, zwei oder mehreren Doppelbindungen. In Abhängigkeit von der relativen Anordnung zweier oder mehrerer Doppelbindungen zueinander unterscheidet man 1) kumulierte Doppelbindungen C=C=C (Kumulene), 2) konjugierte Doppelbindungen C=C-C=C (Diene oder Polyene) und 3) isolierte Doppelbindungen C=C-C-C=C (Diolefine).
A. im engeren Sinne, einfache A., enthalten nur eine Doppelbindung. Sie bilden eine homologe Reihe von Verbindungen der allg. Formel CnH2n, beginnend mit Ethen (Tab.). Erstmals beim Buten treten Konstitutionsisomere und in Abhängigkeit von der Lage der Doppelbindung Konfigurationsisomere auf, z. B. E- und Z-But-2-en (Stereoisomerie).
Alkene. Tab.: Homologe Reihe
| ||
| Propen | CH2=CH-CH3 | |
| But-1-en | CH2=CH-CH2-CH3 | |
| But-2-en | CH3-CH=CH-CH3 | |
| Isobuten, 2-Methylprop-1-en | (CH3)2C=CH2 | |
| Pent-1-en | CH2=CH-CH2-CH2-CH3 | |
| Hex-1-en | CH2=CH-CH2-CH2-CH2-CH3 |
Über die Bezeichnung der A. Nomenklatur Abschn. C III.
Eigenschaften. A. unterscheiden sich bezüglich der physikalischen Eigenschaften kaum von den analogen Alkanen, d. h., in der homologen Reihe gibt es gasförmige (C2 bis C4), flüssige (C5 bis C17) und feste Vertreter. Von den E,Z-isomeren A. hat im allgemeinen das E-Diastereomere einen höheren Schmelzpunkt, einen tieferen Siedepunkt und einen niedrigeren Brechungsindex als das Z-Diastereomere.
A. sind in Wasser schwer löslich, jedoch löslich in Ether und Ethanol. Sie sind brennbar und verbrennen im allg. zu Kohlendioxid und Wasser, bei Sauerstoffmangel auch zu Kohlenmonoxid und Ruß.
Im Gegensatz zu Alkanen sind A. außerordentlich reaktionsfähig.
Reaktionen. Typisch für A. sind Additionsreaktionen, da das chemische Verhalten der A. weitgehend durch die π-Bindung mit ihrer relativ niedrigen π-Bindungsenergie bestimmt wird. Da die C=C-Doppelbindung nucleophilen Charakter hat, sind neben radikalischen Additionen elektrophile Umsetzungen bevorzugt: 1) Katalytische Hydrierung zu Alkanen, die bei Zimmertemperatur und Normaldruck oder leichtem Überdruck (im Labor), in der Technik bei erhöhten Temperaturen und höheren Drücken erfolgt:
Diese Umsetzung dient in Technik und Labor sowohl zur Synthese gesättigter Verbindungen aus ungesättigten als auch zur quantitativen Bestimmung der Anzahl von Doppelbindungen in Molekülen.
2) Addition von Halogenen, die bei Zimmertemperatur verläuft, wobei 1,2-Addukte gebildet werden: R-CH=CH-R + Br2 → R-CH(Br)-CH(Br)-R. Besondere Bedeutung hat diese Umsetzung zum qualitativen Nachweis einer C=C-Doppelbindung, z. B. durch Entfärbung einer verd. Bromlösung beim Zutropfen zu einer Probelösung, oder zur quantitativen Bestimmung der Anzahl der Doppelbindungen im Molekül, z. B. bei der Bestimmung der Iodzahl von Fetten (Addition von Iod an die Doppelbindungen in Fettmolekülen). Die Entfärbung beruht dabei auf der Bildung der nahezu farblosen 1,2-Addukte unter Verbrauch von Brom oder Iod. Man kann auch mit einem Überschuß an Iod arbeiten und die nicht zur Addition verbrauchte Menge Iod quantitativ durch iodometrische Titration bestimmen.
3) Addition von Halogenwasserstoffen, die zu Halogenalkanen führt: CH2=CH2 + HBr → CH3-CH2-Br. Die Reaktivität der A. gegenüber Halogenwasserstoffen steigt in Richtung zunehmender Säurestärke an: HF < HCl < HBr < HI. Bei unsymmetrisch substituierten A. erfolgt die elektrophile Addition regioselektiv nach der Markownikoffschen Regel, die besagt, daß das elektronegative Halogenatom an das wasserstoffärmste C-Atom addiert wird: CH3-CH=CH2 + HBr → CH3-CH(Br)-CH3 (2-Brompropan; Markownikoff-Produkt). Durch Zugabe von Peroxiden kann die Reaktion nach Kharasch als radikalische Addition ablaufen, wobei bevorzugt das anti-Markownikoff-Produkt entsteht, z. B. 1-Brompropan aus Propen:
4) Hydratisierung, d. h. Addition von Wasser unter Bildung von Alkoholen, die Umkehr der Bildung von A. aus Alkoholen (Dehydratisierung). Sie gelingt mittels Säurekatalyse, folgt der Markownikoffschen Regel und ist z. B. für die Gewinnung von tert-Butanol (Butanole) technisch wichtig. Bei Verwendung von Schwefelsäure wird diese zunächst addiert, wobei saure Schwefelsäureester (Alkylsulfate) entstehen, die als Natriumsalze Waschrohstoffe (Tenside) sind. Die Hydrolyse der Alkylsulfate führt ebenfalls zu Alkoholen, z. B.:
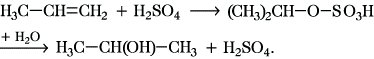
Andererseits können Alkylsulfate mit einem Überschuß an Alkohol leicht zu Ethern umgesetzt werden.
5) Hydroxylierung zu 1,2-Diolen (Glycole), die mit einer verd. alkalischen Lösung von Kaliumpermanganat unter Entfärbung und Abscheidung von Braunstein gelingt. Diese Reaktion kann zum Nachweis der Alkendoppelbindung dienen (Baeyersche Probe):

Die Hydroxylierung ist im Labormaßstab auch mit Osmium(VIII)-oxid oder dem weniger toxischen Ruthenium(VIII)-oxid möglich. Sie erfolgt im Sinne einer Additionsreaktion in Gegenwart von Wasser und den genannten Oxidationsmitteln, wobei als cyclische Intermediate Glycolester von Mangan-, Osmium- und Rutheniumsauerstoffsäuren entstehen, die dann hydrolysiert werden:
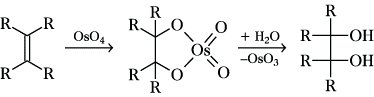
6) Epoxidierung, eine zu Epoxiden führende Addition von Sauerstoff an die Alkendoppelbindung:
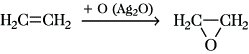
So entsteht z. B. aus Ethen mit Silberoxid Ethylenoxid (Oxiran). Weitere mögliche Reagenzien sind Persäuren, wie Perbenzoesäure und Monoperphthalsäure, oder auch Hydroperoxide in Gegenwart von Molybdän- und Wolframkatalysatoren. Oxirane können säurekatalysiert unter Ringöffnung in Diole umgewandelt werden.
7) Carbonylierung, eine Umsetzung von A. mit Kohlenmonoxid und Wasser in Gegenwart von Nickeltetracarbonyl bei 250 °C und Drücken von 2·104 kPa zu Carbonsäuren. Auch saure Katalysatoren sind unter veränderten Reaktionsbedingungen einsetzbar:
8) Carbenübertragung, eine Addition von Carbenen an die Alkendoppelbindung unter Bildung von Cyclopropanderivaten, z. B. im Verlauf einer Simmons-Smith-Reaktion. Besonders leicht wird 1,1-Dichlorcarben aus Chloroform gewonnen und addiert, z. B. an Cyclohexen:
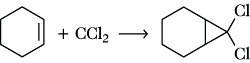
Neben den einfachen Additionsreaktionen neigen A. zur Polymerisation, d. h. zur Polyaddition vieler Moleküle (Monomere) aneinander unter Ausbildung von Makromolekülen (Polymere):
Neben Polyethylen und Polypropylen sind Polyvinylchlorid (PVC), Polystyrol, Polyacrylnitril, Polyacrylester u. a. von großer technischer Bedeutung.
A. gehen auch eine außergewöhnliche Substitution (Allylsubstitution) ein, die unter radikalischen Bedingungen verläuft. So bildet sich z. B. aus Propen und Chlor bei 400 bis 600 °C Allylchlorid: CH2=CH-CH3+ Cl2 → CH2=CH-CH2-Cl + HCl. Eine Allylsubstitution, die z. B. vom Cyclohexen zu 3-Bromcyclohexen führt, ist die Umsetzung mit N-Bromsuccinimid in Gegenwart von Peroxiden oder bei UV-Lichtbestrahlung (Wohl-Ziegler-Reaktion).
Analytisches.Der Nachweis der Doppelbindungen erfolgt mit alkalischer Permanganatlösung (Baeyersche Probe) oder mit Bromlösung in Chloroform, wobei beide Lösungen entfärbt werden. Zur Strukturaufklärung werden vor allem spektroskopische Methoden eingesetzt: In den IR-Spektren finden sich starke Banden im Bereich der C=C-Valenzschwingungen zwischen 1620 und 1680 cm-1. Die C-H-Valenzschwingungen erscheinen bei Wellenzahlen von 3010 bis 3095 cm-1. E,Z-isomere A. haben verschiedene C-H-Deformationsschwingungen: E = 960 bis 980 cm-1, Z = 650 bis 720 cm-1. In den UV-Spektren treten Absorptionsbanden zwischen 180 und 200 nm auf. In den 1H-Kernresonanzspektren liegen die Signale für olefinische Protonen bei δ = 4,3 bis 6 ppm und unterscheiden sich damit charakteristisch von anderen Kohlenwasserstoffen. Der Nachweis für die E- oder Z-Konfiguration ist oft leicht mittels der vicinalen Kopplungskonstanten J zu führen: JZ = 5 bis 16 Hz; JE = 13 bis 21 Hz. Das massenspektrometrische Fragmentierungsverhalten der A. ist durch eine Allylspaltung des Molekül-Ions gekennzeichnet, wobei die gebildeten Allyl-Kationen mesomeriestabilisiert sind. Eine Bestimmung der Position der C=C-Doppelbindung ist mittels der Massenspektrometrie nicht möglich.
Herstellung. Niedere A. (C2 bis C5) werden im technischen Maßstab aus den bei der Erdölverarbeitung anfallenden Raffineriegasen abgetrennt oder durch katalytische Dehydrierung von Alkanen oder Pyrolyse von Benzin oder Kerosin bei 800 bis 900 °C gewonnen. Daneben gibt es zahlreiche charakteristische Synthesemethoden für den Laboratoriumsmaßstab oder für wissenschaftliche Zwecke:
1) Dehydratisierung von Alkoholen durch Erhitzen mit Schwefelsäure, Phosphorsäure oder Zinkchlorid:
Von größerer Bedeutung ist die katalytische Dehydratisierung an Aluminiumoxid- oder Thoriumoxid-Katalysatoren in der Gasphase, bei der Konkurrenzreaktionen, z. B. die Bildung von Ethern, weitgehend unterbunden werden können.
2) Dehydrohalogenierung von Halogenalkanen mit Basen: CH3-CH2-Br + KOH → CH2=CH2 + KBr + H2O.
3) Dehalogenierung von 1,2-Dihalogenalkanen durch Einwirkung von Zink, Natriumiodid in Methanol, Chrom(II)-Salzen oder von Natriumthiosulfat in Dimethylsulfoxid:
4) Einführung einer Doppelbindung durch Hofmann-Abbau, ausgehend von quartären Ammoniumhydroxiden.
5) Einführung einer Doppelbindung durch Cope-Reaktion, ausgehend von tert-Aminoxiden.
6) Umsetzung von Aldehyden oder Ketonen mit Alkylidenphosphoranen im Sinne der Wittig-Reaktion.
7) Reduzierende Kupplung von Aldehyden oder Ketonen durch Behandlung mit Lithiumaluminiumhydrid: 2 (R)2C=O → (R)2C=C(R)2.
8) Pyrolyse von Carbonsäureestern bei Temperaturen von etwa 500 °C, wobei eine synchrone cis-Eliminierung abläuft:
;)
9) Pyrolyse von Xanthogensäureestern (Tschugaeff-Reaktion) bei etwa 200 °C, ebenfalls als synchrone cis-Eliminierung:
;)
Verwendung. In großtechnischem Maßstab bei petrolchemischen Prozessen gewonnen, werden A. vorrangig zur Herstellung von hochverzweigten Alkanen, von Alkoholen, Ethern, Epoxiden und Glycolen, Carbonsäuren, Halogenalkanen, Alkylbenzolen, Alkylsulfaten und synthetischen Polymeren eingesetzt.







Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.