Lexikon der Chemie: elementorganische Verbindungen
elementorganische Verbindungen. Organoelementverbindungen, Elementorganyle, Verbindungen, die über eine oder mehrere direkte Element-Kohlenstoff-Bindungen verfügen. Die Bindungsart ist dabei ohne Belang (Verbindungen mit Element-Kohlenstoff-σ-Bindung, π-Olefin- (Alken-) und π-Aromatenkomplexe u. ä.). Kohlenstoffderivate der Halogene, des Sauerstoffs, des Schwefels und des Stickstoffs werden ausgeklammert und traditionsgemäß der organischen Chemie zugeordnet. Ist das zugrunde liegende Element ein Metall, spricht man von metallorganischen Verbindungen (Organometallverbindungen). Nicht zur Gruppe der e. V. gehören Derivate, in denen der organische Ligand nicht über ein Kohlenstoffatom, sondern über z. B. Sauerstoff oder Stickstoff an das Element gebunden ist, d. h. Salze von Carbonsäuren, Alkoxide, Amide oder Ester der Phosphorsäure und ähnliche Verbindungen.
Einteilung. Auf der Grundlage der bei der Vielzahl von e. V. naturgemäß sehr unterschiedlichen Bindungstypen hat sich folgende Klassifizierung bewährt:
1) ionische e. V., die Derivate der elektropositivsten Elemente, insbesondere der schwereren Alkali- und Erdalkalimetalle Natrium, Kalium, Rubidium, Cäsium bzw. Calcium, Strontium und Barium, meist schwerflüchtige, in Kohlenwasserstoff unlösliche, extrem reaktive Verbindungen. Typische Vertreter sind Ethinylnatrium (Natriumhydrogenacetylid) Na+-C≡CH und Methylkalium (Kaliummethanid), K+CH3- (Nickelarsenidtyp).
2) kovalente e. V., für die lokalisierte 2-Zentren-2-Elektronen-Bindungen typisch sind. Sie werden von den weniger elektropositiven Elementen Gallium, Indium, Thallium, den Elementen der IV., V. und VI. Hauptgruppe sowie von Zink, Cadmium und Quecksilber gebildet. Es sind meist monomere, leichtflüchtige, in Kohlenwasserstoffen lösliche Verbindungen. Beispiele: Bortrialkyle R3B, Dibutylzinndichlorid (C4H9)2SnCl2, Ethanphosphonsäure C2H5P(O)(OH)2, Diphenylquecksilber (C6H5)2Hg.
3) e.V. mit Mehrzentrenbindungen ("Elektronenmangelstrukturen"), speziell die Organoderivate des Berylliums, Magnesiums und Aluminiums, in gewissem Umfange auch die des Lithiums und des Bors. Die meist dimeren, oligomeren oder polymeren Strukturen dieser Verbindungen werden durch die Ausbildung von 3-Zentren-2-Elektronen-Bindungen bestimmt, wodurch die Elektronenkonfigurationen der Zentralatome die eines Edelgases erreichen oder sich dieser zumindest annähern. Typische Beispiele: Methyllithium (CH3Li)4 (in Festsubstanz), Dimethylberyllium [(CH3)2Be]n und die Aluminiumtrialkyle (R3Al)2.
Zwischen diesen drei Verbindungsgruppen treten Überschneidungen auf, z. B. bei Lithium (ionisch/ Elektronenmangel) oder Bor (Elektronenmangel/ kovalent).
4) Übergangsmetallorganyle. Während die vorstehende Kategorisierung der Organoderivate der Hauptgruppenelemente auf der Basis üblicher Bindungsvorstellungen aus der Stellung dieser Elemente im Periodensystem deutlich wird, sind für das Verständnis der Übergangsmetallorganyle vor allem koordinationschemische und bindungstheoretische Gesichtspunkte zu berücksichtigen, und man nimmt eine weitere Unterteilung dieser Verbindungen vorteilhaft nach dem Bindungstyp (σ-Donor-, σ-Donor-/ π-Akzeptor- oder σ,π-Donor-/π-Akzeptor-Liganden) und der Art des Liganden (z. B. Alkyl-, Alkyliden-, Allyl-, Alken-, Aren-Komplexe u. a.) vor. Das rechtfertigt eine besondere Behandlung dieser Verbindungsklasse. Dem sei jedoch zunächst eine zusammenfassende Diskussion des Verhaltens der erstgenannten drei Verbindungstypen, die sämtlich von Hauptgruppenelementen gebildet werden, vorangestellt.
e. V. der Hauptgruppenelemente (einschließlich der Derivate des Zinks, Cadmiums und Quecksilbers). Stabilität und Reaktivität. Bei vergleichbar gebauten Verbindungen der Hauptgruppenelemente nimmt die thermische Stabilität mit steigender Ordnungszahl des Elements üblicherweise ab. Das chem. Verhalten wird durch die meist stark im Sinne von Eδ+-Cδ- polarisierte Element-Kohlenstoff-Bindung oder die im Falle ionischer Verbindungen mehr oder weniger frei vorliegenden Carbanionen bestimmt. Metallorganica sind deshalb meist kraftvolle Nucleophile und starke Basen. Dabei wird die Reaktivität in erster Linie von der Polarität der Metall-Kohlenstoff-Bindung, d. h. der Elektronegativität des Elements, bestimmt. Lithiumorganische Verbindungen sind deshalb im allg. reaktiver als Grignard-Reagenzien. Daneben ist jedoch auch die Natur des organischen Restes von Bedeutung, was besonders beim Vergleich der Reaktivitäten verschiedener Organoderivate desselben Elements deutlich wird. So steigt die Beständigkeit bzw. sinkt die Reaktivität ionischer e. V. mit zunehmender Stabilität des Carbanions oder – gleichbedeutend – mit steigender Acidität des diesem zugrunde liegenden Kohlenwasserstoffs. In kovalenten e. V. wird die Reaktivität in starkem Maße vom Lösungsmittel beeinflußt, das im Falle guter Donorfähigkeit das positivierte Metall koordiniert, dessen Elektronegativität verringert, damit die Bindungspolarität und und so die nucleophile Kraft des Kohlenstoffs erhöht. dies begründet, weshalb z. B. lithiumorganische Verbindungen in Tetrahydrofuran oder Diethylether reaktiver sind als in Benzol oder aliphatischen Kohlenwasserstoffen. Mit abnehmender Elektronegativitätsdifferenz zwischen Kohlenstoff und dem betreffenden Element nimmt die Beständigkeit der Verbindungen zu, was sich z. B. darin ausdrückt, daß die Bindung der Elemente der IV. und V. Hauptgruppe zu Kohlenstoff durch Luft und Wasser meist nicht gespalten wird.
Typische Verhaltensweisen metallorganischer Verbindungen sind die oft hohe Oxidationsempfindlichkeit und die leichte Hydrolysierbarkeit der Metall-Kohlenstoff-Bindung. So entflammen zahlreiche Alkylderivate unter anderem des Bors, Aluminiums, der Alkali- und Erdalkalimetalle und des Zinks an der Luft spontan und können deshalb nur unter Inertgasatmosphäre gehandhabt werden. Diese reaktiven metallorganischen Verbindungen werden durch Wasser oft explosionsartig zu den entsprechenden Kohlenwasserstoffen und dem Metallhydroxid hydrolysiert, z. B. C4H9-Li +H2O → C4H10 + LiOH. Verbindungen mit stark polarisierten Metall-Kohlenstoff-Bindungen, z. B. Organyle des Lithiums, Magnesiums und z. T. des Aluminiums, vermögen mit geeigneten C=C-, C=O-, und C=N-Doppelbindungssystemen unter Insertion zu reagieren, z. B. C6H5-MgBr + O=C=O → C6H5-COO- MgBr+, und stellen damit wertvolle Bausteine für die organische Synthese dar (z. B. Grignard-Reaktionen, Reformatski-Reaktion).
Synthese. Einige ionische e. V. lassen sich durch unmittelbare Einwirkung des Metalls auf besonders acide Kohlenwasserstoffe herstellen, z. B. Na + C6H5-C≡CH → C6H5-C≡C- Na+ + 1/2 H2 (Metallierung). Auch durch Umsetzung elektropositiver Metalle mit relativ leicht zugänglichen quecksilberorganischen Verbindungen sind entsprechende Metallorganica darstellbar, z. B. C6H5-Hg-C6H5 + 2 Li → 2 C6H5-Li + Hg (Transmetallierung). Von herausragender Bedeutung ist die Synthese aus Alkyl- oder Arylhalogenid und dem Metall, die insbesondere zur Gewinnung von lithium- und magnesiumorganischen Verbindungen genutzt wird, z. B. CH3-I + 2 Li → CH3Li + LiI; C6H5-Br + Mg → C6H5-MgBr (Direktsynthese). Letztere sind sehr häufig Ausgangsmaterial zur Synthese anderer e. V., speziell der Organoderivate der Elemente der III. bis VI. Hauptgruppe sowie der Übergangsmetalle, indem diese mit entsprechenden Elementhalogeniden zur Reaktion gebracht werden. Unter gleichzeitiger Bildung des Metallhalogenids wird das Element alkyliert oder aryliert, z. B. C6H5-MgBr + (CH3)2SiCl2 → (CH3)2C6H5SiCl + MgBrCl; C4H9-Li + (C6H5)2BCl → (C6H5)2B-C4H9 + LiCl; CrCl3 + 3 C6H5MgBr (in THF) → (C6H5)3Cr(THF)3 + 3 MgBrCl. Zahlreiche e. V. sind durch Addition von Organoelementhydriden an C=C-Doppelbindungssysteme zugänglich. Diese Verfahrensweise hat insbesondere bei der Synthese bororganischer Verbindungen (Hydroborierung) und siliciumorganischer Verbindungen (Hydrosilylierung) sowie in der Chemie phosphororganischer Verbindungen Bedeutung, z. B.:
(CH3)2C=CH2 + H-BR2 → (CH3)2CH-CH2-BR2;
(C2H5O)2P(O)H + CH2=CHCN →
(C2H5O)2P(O)CH2CH2CN.
Verwendung. Lithium- und magnesiumorganische Verbindungen sind unentbehrliche Hilfsmittel in der organischen Synthese (Grignard-Reaktionen), ebenso zink- und quecksilberorganische Verbindungen (Reformatski-Reaktion). Phosphororganische Verbindungen in Form der P-Ylide dienen zur Carbonylolefinierung (Wittig-Reaktion). Von herausragendem technischen Interesse sind phosphororganische Verbindungen als Biozide, Flammenschutzmittel, Detergenzien, Schmieröladditive u. a., siliciumorganische Verbindungen vor allem als Polysiloxane (Silicone), zinnorganische Verbindungen als Biozide und PVC-Stabilisatoren, Tetraethylblei als Antiklopfmittel (stark rückläufige Bedeutung) und Aluminiumalkyle zur Alkenoligomerisierung und Gewinnung von 1-Alkenen und 1-Alkanolen sowie als Bestandteil von Katalysatorsystemen zur Alkenpolymerisation (Ziegler-Natta-Niederdruckverfahren). Detaillierte Informationen zu einzelnen Verbindungsklassen z. B. bei aluminiumorganischen Verbindungen, arsenorganischen Verbindungen usw.
Übergangsmetallorganyle. In den Organoderivaten der Übergangselemente kann der Kohlenstoffligand sowohl über σ- als auch π-Beziehungen an das Metallatom gebunden sein. Die Stabilität der Verbindungen ist stark von der Elektronenkonfiguration des zentralen Metallatoms abhängig und kann durch Koordination weiterer geeigneter Liganden beeinflußt werden. Als empirische Grundregel gilt, daß stabile Komplexe zu erwarten sind, wenn die Summe der die jeweiligen s-, p- und D-Orbitale des Metalls besetzenden Elektronen 18 beträgt, d. h. die nächste Edelgaskonfiguration erreicht wird (18-Elektronenregel). Die Elektronenanzahl, mit der – als neutral angenommene – organische Liganden zur Komplettierung der Elektronenhülle beitragen, geht aus der Tab. hervor.
Bei Übergangsmetallkomplexen von Polyenen, Aromaten u. ä. ist allein die Angabe des organischen Liganden zur genaueren Beschreibung der Struktur der Verbindung unzureichend, weil durchaus nicht alle π-Bindungselektronen mit dem Metallatom in Wechselwirkung stehen müssen. Deshalb wurde das hapto-System eingeführt (griech. haptein ›befestigen, verbinden‹), wobei durch Voranstellen des hapto-Symbols η, versehen mit einem Exponenten (η1, η2, η3, usw.) die Anzahl der mit dem Zentral-Ion unmittelbar verknüpften Atome des Liganden angegeben wird. So vermag z. B. die Allylgruppierung als σ-gebundener monohapto-, selten jedoch auch als σ- und π-gebundener mono- und dihapto- sowie meist als π-gebundener trihapto-Ligand (Abb. 1) zu fungieren. Ein Komplex, in dem zwei verschiedene Bindungsarten des Cyclopentadienylliganden realisiert sind, ist z. B. das Dicarbonyl(η1-cyclopentadienyl)(η5-cyclopentadienyl)eisen:
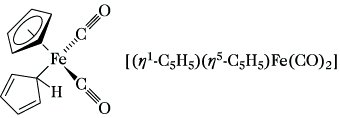
elementorganische Verbindungen. Tab.: Klassifikation neutral gerechneter organischer Gruppen als Liganden in Übergangsmetallorganylen.
| |||
| 1 | -yl | Methyl, σ-Allyl, Phenyl | |
| 2 | -en | Ethen, Tetrafluorethen | |
| 3 | -enyl | η3-Allyl, η3-Cyclopropenyl | |
| 4 | -dien | Butadien, Cyclobutadien | |
| 5 | -dienyl | η5-Cyclopentadienyl | |
| 6 | -trien | Benzol, η6-1,5,9-Cyclododecatrien | |
| 7 | -trienyl | η7-Cycloheptatrienyl | |
| 8 | -tetraen | η8-Cyclooctatetraen |
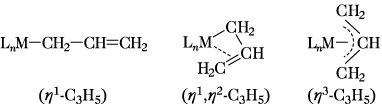
elementorganische Verbindungen. Abb. 1: Bindungsmöglichkeiten der Allylgruppe.
Struktur und Synthese. 1) Einelektronenliganden. Die thermische Beständigkeit von Organylen des Typs LnMRm (R = Alkyl, Aryl; L = CO, PR3 u.a.), in denen die Gruppe R als σ-Donorligand bindet, variiert in weiten Grenzen. Homoleptische Methylderivate MMen sind entweder in Substanz nicht darstellbar (z. B. NiMe2) oder sehr labil (z. B. TiMe4: Zersetzung bereits ab -70 °C), jedoch zeigen Vertreter mit koordinativ gesättigtem Metallzentrum relativ inertes Verhalten (z. B. WMe6, rote Kristalle, F. 30 °C). Die hohe Zersetzlichkeit vieler Übergangsmetallalkyle geht jedoch nicht auf geringe thermodynamische Stabilität zurück, sondern hat kinetische Ursachen. So ist die thermische Labilität längere Alkylgruppen enthaltender Derivate vor allem auf die unter β-Hydridverschiebung verlaufende rasche Olefinabspaltung (reduktive Eliminierung) zurückzuführen (Abb. 2). Wird diese β-Eliminierung dadurch unterdrückt, daß die Gruppe R kein β-H-Atom aufweist (z. B. R = Benzyl) oder die Abspaltung des Olefins ungünstig ist (z. B. bei R = Norbornyl), werden thermisch recht beständige Verbindungen erhalten. So zersetzt sich Tetramethylzirconium bereits ab -15 °C, während das Benzylderivat ZrBz4 bis zum Schmelzpunkt bei 131 °C stabil ist. Eine deutlich erhöhte Stabilität wird auch durch den Einbau von Stützliganden erreicht (Beispiel: [(CO)4Co-CF3] ist bei 91 °C destillierbar). Die sich teilweise explosionsartig zersetzenden homoleptischen Alkinylmetallate (z. B. [Ni(C≡CPh)4]2-, [Cr(C≡CCH3)6]3-) sind in vielen Eigenschaften den Cyanometallaten vergleichbar (CN- und HC≡C- sind isoelektronisch). Mit zusätzlichen Liganden gelingt auch hier eine deutliche Stabilisierung. So ist das orangegelbe trans-[Ni(C≡CPh)2(PEt3)2] bis zum Schmelzpunkt von 149 °C beständig.
Man gewinnt σ-Organoübergangsmetallverbindungen durch Alkylierung oder Arylierung geeigneter Koordinationsverbindungen mit reaktiven Metallorganylen: [(Et3P)2PtCl2] + CH3MgI → [(Et3P)2Pt(CH3)I] + MgCl2; [(Et3P)2PtCl2] + 2 CH3-Li → [(Et3P)2-Pt(CH3)2] + 2 LiCl. Eine weitere Verfahrensweise besteht in der Umsetzung von Carbonylmetallaten der Übergangsmetalle mit Alkylhalogeniden, z. B. Na+[Mn(CO)5]- + CH3I → [(CO)5MnCH3] + NaI.
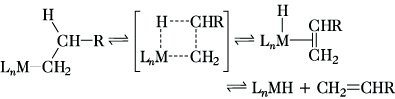
elementorganische Verbindungen. Abb. 2: Thermische Zersetzung von Übergangsmetallalkylen.
2) Carbenkomplexe (Alkylidenkomplexe). Organyle der allg. Zusammensetzung LnM=C(X)(Y) (L = CO, PR3, H, R u. a.) lassen sich mit Metallen der IV.-VIII. Nebengruppe synthetisieren, in denen Carbene des Typs :C(X)(Y) (X, Y = H, R, OR, NR2 u. a.) als σ-Donor-/π-Akzeptorliganden unter Lieferung von zwei Elektronen und Ausbildung einer M=C-Doppelbindung koordiniert sind. Dabei liegt in den 1964 erstmals von E. O. Fischer und A. Maasböl erhaltenen "Fischer-Carbenkomplexen" das Metall in niedriger Oxidationsstufe vor und das Heteroatom-substituierte Carben-C-Atom reagiert elektrophil. Dagegen weisen die erstmals 1974 von R. Schrock synthetisierten, wesentlich reaktiveren "Schrock-Carbenkomplexe" Metalle in höheren Oxidationsstufen auf, das koordinierte Carben enthält keine Heteroatome (X, Y = H, R) und zeigt nucleophiles Verhalten. C. des Fischer-Typs lassen sich z. B. durch Alkylierung von Metallhexacarbonylen mit Lithiumalkylen und nachfolgende Methylierung des intermediär entstehenden Anions herstellen:
;)
C. des Schrock-Typs können z. B. durch α-Deprotonierung einer M-Alkylgruppe erhalten werden: [(η5-C5H5)2Ta(CH3)2]+ + CH3O- → [(η5-C5H5)2(CH3)Ta=CH2] + CH3OH. Carbenkomplexe spielen eine wichtige Rolle in der organischen Synthese, als Methylenübertragungreagenzien (Tebbe-Reagens, titanorganische Verbindungen) und bei der homogenkatalysierten Alken-Metathese.
3) Carbinkomplexe (Alkylidinkomplexe). Von Übergangsmetallen der V.-VII. Nebengruppe sowie der Eisengruppe (Gruppe 8) sind Organyle des allgem. Typs LnM≡CR (L = CO, PR3, η5-C5H5, R u.a.) bekannt, in denen die Carbingruppe ·C·· (R = Alkyl, Aryl) als Dreielektronenligand mit σ-Donor-/π-Akzeptorcharakter unter Ausbildung einer M≡C-Dreifachbindung koordiniert. Die erstmals 1973 von E. O. Fischer und U. Schubert erhaltenen Carbinkomplexe lassen sich z. B. ausgehend von Carbenkomplexen durch Reaktion mit Bortrihalogeniden gemäß
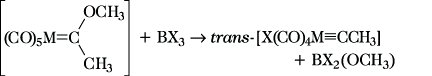
oder durch α-Deprotonierung entsprechend
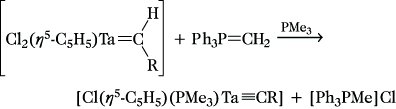
erhalten.
In den vorstehend behandelten Übergangsmetallorganylen wird ein freies Elektronenpaar am C-Atom der Gruppe R zur Knüpfung der M-C-σ-Bindung verwendet. Besondere Bedeutung kommt weiterhin den im folgenden beschriebenen π-Komplexen zu, bei denen der C-Ligand (mindestens) ein Elektronenpaar, das sich in einem Orbital mit π-Symmetrie hinsichtlich der intra-Ligand-Bindung befindet, für die Koordination an das Metall zur Verfügung stellt.
4) Olefinkomplexe. Die Synthese gelingt am einfachsten durch Substitution geeigneter Liganden durch das entsprechende Olefin. So kann der bereits 1827 als erstes Übergangsmetallorganyl beschriebene Platin-Olefin-Komplex K[PtCl3(C2H4)]·H2O, das Zeisesche Salz (platinorganische Verbindungen), präparativ günstig durch Einwirkung von Ethen auf Kaliumtetrachoroplatinat(II) gewonnen werden. Als besonders geeignet zu derlei Austauschreaktionen haben sich Metallcarbonyle erwiesen, z. B. [Fe(CO)5] + Butadien → [(CO)3Fe(CH2= CHCH=CH2)] + 2 CO. Die Bindung des Olefins an das Metall versteht man als dative σ-Wechselwirkung des bindenden π-Elektronenpaares mit einem Metall-Atomorbital geeigneter Symmetrie und eine gleichzeitige π-Rückbindung (π-back-donation) vom Metall über gefüllte D-Niveaus mit π-Symmetrie in antibindende π*-Molekülorbitale des Olefins (Abb. 3). Olefine mit zwei oder mehr isolierten Doppelbindungen werden in analoger Weise an das Metall geknüpft. An der Bindung konjugierter Diene, z. B. des 4-Elektronenliganden Butadien an das Eisen (s. o.), sind delokalisierte π-Molekülorbitale beteiligt.
;)
elementorganische Verbindungen. Abb. 3: Schematische Darstellung der Bindung zwischen Ethen und einem Übergangsmetall-Ion.
5) Allylkomplexe. Stellvertretend sei das η3-Allyltricarbonylcobalt (Abb.4a) genannt, das durch Einwirkung von Allylbromid auf Na+[Co(CO)4]- über den σ-Allylkomplex [(CO)4Co-CH2CH=CH2] gebildet wird. Letzterer geht unter Eliminierung von CO in die π-Allylverbindung über. Als weiteres Beispiel diene das Bis(η3-allyl)nickel (Abb. 4b), das man durch Umsetzung von Nickel(II)-chlorid mit Allylmagnesiumbromid gewinnt. Die C-C-Abstände in den Allylgruppen sind gleich, die H-Atome liegen in der Ebene der Allyl-Kohlenstoffatome. Die Fähigkeit des Allylrestes, sowohl als σ-gebundener Einelektronenligand wie als π-gebundener Dreielektronenligand zu fungieren, ermöglicht unter geeigneten Bedingungen rasche σ,π-Umwandlungen (σ,π-Äquilibrierung), die die Grundlage einiger technischer Prozesse bilden.
;)
elementorganische Verbindungen. Abb. 4: Beispiele für Allylkomplexe. (a) η3-Allyltricarbonylcobalt, (b) Bis(η3-allyl)nickel.
Cyclische ungesättigte Kohlenwasserstoffliganden, die ein konjugiertes System aus (4n + 2) π-Elektronen aufweisen, bilden mit Übergangsmetallen meist besonders stabile Aromatenkomplexe. Hierzu gehören die 6π-Elektronensysteme Benzol bzw. dessen Derivate C6R6, die Cyclopentadienylliganden C5R5- (R = H, Me, Ph u. a.) und das Tropyliumkation C7H7+ sowie der 10π-Elektronenligand C8H82-.
6) Cyclopentadienylkomplexe. Unter den Aromatenkomplexen stellen die Vertreter mit Cylcopentadienylliganden die wichtigste Stoffklasse dar. Von nahezu allen Übergangsmetallen sind homoleptische Komplexe der Zusammensetzung [M(η5-C5H5)2] (Metallocene) und weiterhin, mit einigen Metallen, des allgemeinen Typs [M(η5-C5H5)n] (z. B. n = 4: M = Ti, Zr, Hf, U), sowie gemischte, noch zusätzliche Liganden enthaltende Verbindungen bekannt. Repräsentativer Vertreter für die Sandwichstruktur aufweisenden Metallocene ist das Ferrocen (Abb. 5a). Auch Mehrfachdecker-Sandwichsysteme, z. B. das Tripeldecker-Komplexkation [Ni2(C5H5)3]+ (Abb. 5b) lassen sich erhalten.
Typische Beispiele für Bis (η5-cyclopentadienyl)-Derivate, deren C5H5-Ringe nicht parallel stehen ("gewinkelte Metallocene") stellen das Dichlorobis(η5-cyclopentadienyl)titan(IV) (Abb. 5c), für Halbsandwichkomplexe das Tricarbonyl(η5-cyclopentadienyl)mangan(I) (Abb. 5d) dar. Man interpretiert die Bindung des C5R5-Liganden an das Übergangsmetall-Ion am besten mit Hilfe der Molekülorbitaltheorie, wobei die Molekülorbitale des Komplexes durch Kombination der π-MOs des cyclopentadienyl-Anions mit Metallorbitalen entsprechender Symmetrie gebildet werden. Die thermisch oft bemerkenswert stabilen Verbindungen ermöglichen Reaktionen am C5H5-Ring, die auf dessen aromatischen Charakter hinweisen.
Man erhält Metallocene meist durch Reaktion von Metallhalogeniden mit Natrium-cyclopentadienid, z. B. FeCl2 + 2 NaC5H5 → [η5-C5H5)2 Fe] + 2 NaCl.
7) Arenkomplexe. Die 6π-Elektronensysteme Benzol bzw. dessen Derivate C6R6 sowie kondensierte Sechsringaromaten wie Naphthalin, Phenanthren, Coronen bilden mit vielen Übergangsmetallen homoleptische einkernige Aromatenkomplexe mit Sandwichstruktur, in denen diese Liganden jeweils über einen Sechsring η6-gebunden sind.
Typischer Vertreter ist das Bis-(η6-benzol)chrom(0) (Abb. 6a), in dem die beiden Arenringe planar und parallel zueinander angeordnet sind. Alle C-C und C-Cr-Abstände sind gleich. Nach der Synthese von E. O. Fischer und W. Hafner wird es aus Chrom(III)-chlorid durch Umsetzung mit Aluminium in Gegenwart von Aluminiumchlorid und Benzol und anschließende Reduktion des zunächst gebildeten Komplexkations [Cr(η6-C6H6)2]+ mit Dithionit erhalten. Auch Gemischtligandkomplexe wie (η6-Benzol)tricarbonylchrom(0), [Cr(η6-C6H6)(CO)3], mit Halbsandwichstruktur sind bekannt.
Ein typischer Vertreter für Sandwichkomplexe der Lanthanoide und Actinoide mit dem 10π-Elektronenliganden C8H82- ist das gemäß
erhältliche blaßgrüne, paramagnetische Bis(η8-1,3,5,7-cyclooctatetraen)uran (Uranocen), [U(η8-C8H8)2] (Abb. 6b).
Verwendung. Übergangsmetallorganyle haben zur Aktivierung von Olefinen in zahlreichen technisch wichtigen Synthesen außerordentliche Bedeutung erlangt; sie sind Bestandteil von Katalysatorsystemen zu deren Hydrierung, Polymerisation. Oxidation und Oxosynthese.
;)
elementorganische Verbindungen. Abb. 5: Cyclopentadienylkomplexe: (a) Ferrocen, (b) [Ni2(C5H5)3]+, (c) Dichlorobis(η5-cyclopentadienyl)titan(IV), (d) Tricarbonyl(η5-cyclopentadienyl)mangan(I).
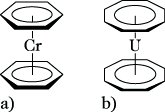
elementorganische Verbindungen. Abb. 6: Arenkomplexe. (a) Bis(η6-benzol)chrom(0) und (b) Uranocen.








Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.