Lexikon der Chemie: Enzyme
Enzyme, früher als Fermente bezeichnet, in der lebenden Zelle gebildete Proteine, die als Biokatalysatoren die chem. Reaktionen des Stoffwechsels beschleunigen und in zunehmendem Maße auch zur selektiven Stoffwandlung (Biotransformation) außerhalb des Zellbereiches eingesetzt werden. Die Beschleunigung ist 103- bis 106fach gegenüber der unkatalysierten Reaktion, die Anzahl der je Enzymmolekül umgesetzten Substratmoleküle kann bis zu 105 je s betragen.
Als Katalysatoren verringern die E. die Aktivierungsenergie der katalysierten Reaktion durch intermediäre Bildung eines Enzym-Substrat-Komplexes. Das thermodynamische Gleichgewicht der Reaktion wird dabei nicht verändert, sondern nur beschleunigt eingestellt. Die E. erscheinen am Ende der Reaktion in ihrer ursprünglichen Form. Das Temperaturoptimum der enzymatisch katalysierten Reaktion liegt bei etwa 50 °C. Oberhalb dieser Temperatur kommt es zur Hitzedenaturierung der Proteinkomponente. Eine Ausnahme bilden die thermophilen E. bestimmter Mikroorganismen, die bis zu 110 °C katalytisch wirksam bleiben. Die Wirkung der E. ist weiterhin vom pH-Wert des Mediums, von der Anwesenheit spezifischer Effektoren sowie von der Substratkonzentration abhängig. Alle E. zeichnen sich durch eine hohe Substrat- und Wirkungsspezifität aus. Generell wird nur eine Verbindung (oder ein Verbindungstyp) als Substrat akzeptiert oder nur ein bestimmter Reaktionstyp beschleunigt, wobei die regio-, stereo- und enantioselektive Umsetzung des Substrats besonders charakteristisch ist. Die volle katalytische Wirksamkeit der E. ist an das Vorliegen einer bestimmten Konformation des Gesamtmoleküls gebunden. Für die Bindung und Umsetzung des Substrats sind dagegen nur einige räumlich benachbarte Aminosäurereste des "aktiven Zentrums" und kooperativ wirksame Effektoren verantwortlich.
Klassifizierung. Die gegenwärtig bekannten mehr als 2500 E. können nach ihrem Vorkommen in der Natur (tierische, pflanzliche, mikrobielle E.), nach ihrer Stellung im Stoffwechsel (Verdauungs-, Atmungsketten-, Blutgerinnungsenzyme), nach ihren funktionellen Gruppen (Serin-, SH-Enzyme), nach ihren physikalischen Eigenschaften und nach vielen anderen Gesichtspunkten eingeteilt werden. Durchgesetzt hat sich jedoch das auf der Wirkungsspezifität beruhende internationale Einteilungssystem (EC-Nomenklatur, Abk. von enzyme commission). Danach erhält jedes E. eine vierstellige Codenummer, die die Hauptgruppe oder Klasse, die Gruppe, die Untergruppe und die Seriennummer festlegt.
Struktureller Aufbau. Als globuläre Proteine können die E. aus nur einer Polypeptidkette bestehen (monomere oder Einkettenenzyme) oder aus mehreren gleichen oder verschiedenen Untereinheiten aufgebaut sein (oligomere oder Mehrkettenenzyme). Einkettenenzyme sind vor allem an das Blut oder an den Verdauungstrakt abgegebene Sekretenzyme. Häufig werden diese E. in Form inaktiver Vorstufen (Zymogene) gebildet und erst am Wirkort durch limitierte Proteolyse freigesetzt. Mehrkettenenzyme gehören zu den Zellenzymen, die nur intrazellulär wirksam werden und vielfach an spezifische Zellstrukturen, z. B. an Membranen, gebunden sind. Allosterische E. sind Mehrkettenenzyme mit Bindungsstellen sowohl für das Substrat als auch für ein Effektor- oder Modulatormolekül (meist das Endprodukt einer Biosynthesekette). Sie dienen der Regulation der Enzymaktivität (Feedback-Regulation, Feedback-Mechanismus). Zu den Mehrkettenenzymen gehören weiterhin die Multienzymkomplexe, bei denen E., die aufeinanderfolgenden Reaktionsschritte katalysieren, als Untereinheiten auftreten und Quartärstrukturen ausbilden, in denen die katalytischen Zentren der Einzelenzyme optimal stabilisiert sind. Im Gegensatz zu den Multienzymkomplexen sind die mehrere katalytische Funktionen ausübenden multifunktionellen E. gewöhnlich Einkettenenzyme. Einige E. üben innerhalb des gleichen Organismus gleiche Funktionen aus. Weist ihr Aufbau eine unterschiedliche Aminosäuresequenz auf, werden sie als Isoenzyme bezeichnet.
Etwa die Hälfte aller Enzyme benötigt außer der Proteinkomponente (Apoenzym) noch Coenzyme und Metall-Ionen als Cofaktoren. Diese sind entweder fester Bestandteil der E. (prosthetische Gruppe) oder werden von den aktiven Formen nur reversibel gebunden. Für das Zustandekommen der katalytischen Wirkung sind Proteinkomponenten und Cofaktoren gemeinsam (Holoenzym) verantwortlich.
| ||
| 1.1 1.2 1.3 1.4 | wirken auf /CHOH wirken auf /C=O wirken auf -CH2-CH2- wirken auf /CH-NH2 | Alkoholdehydrogenase 1.1.1.1 Formiatdehydrogenase 1.2.1.2 Succinatdehydrogenase 1.3.99.3 L-Aminosäureoxidase 1.4.3.2 |
| 2 | Transferasen: katalysieren intermolekulare Gruppenübertragungen | |
| 2.1 2.2 2.3 2.4 2.5 2.6 2.7 | C1-Gruppenübertragung Carbonylgruppen Acylgruppen Glycosylgruppen Alkyl-, Arylgruppen Aminogruppen phosphorhaltige Gruppen | Aspartatcarbamoyl-Transferase 2.1.2.3 Transketolase 2.2.1.1 Cholinacetyltransferase 2.3.1.6 Glycosyltransferasen 2.4 Aminotransferasen 2.6.1 Nucleotidyltransferasen 2.7.7 |
| 3 | Hydrolasen: katalysieren hydrolytische Spaltung von | |
| 3.1 3.2 3.3 3.4 3.5 | Esterbindungen Glycosidbindungen Etherbindungen Peptidbindungen anderen C-N-Bindungen | Lipase 3.1.1.3 α-Amylase 3.2.1.1 Thioetherhydrolasen 3.3.1 Leucinaminopeptidase 3.4.11.1 Urease 3.5.1.5 |
| 4 | Lyasen: katalysieren Eliminierungsreaktionen unter Bildung von Doppelbindungen oder, als Synthasen bezeichnet, Additionen an Doppelbindungen | |
| 4.14.24.3 | C-C-Lyasen C-O-Lyasen C-N-Lyasen | Pyruvatdecarboxylase 4.1.1.1 Carboanhydrase 4.2.1.1 Aspartase 4.3.1.1 |
| 5 | Isomerasen: katalysieren Isomerisierungsreaktionen | |
| 5.15.25.3 5.4 | Racemasen-Epimerasen cis-trans-Isomerasen intramolekulare Oxidoreduktasen intramolekulare Transferasen | Alaninracemase 5.1.1.1 Retinalisomerase 5.2.1.3 Triosephosphatisomerase 5.3.1.1 Phosphotransferasen 5.4.2 |
| 6 | Ligasen (Synthetasen): katalysieren die Verknüpfung zweier Moleküle unter ATP-Verbrauch | |
| 6.16.26.36.4 | C-O-Verknüpfung C-S-Verknüpfung C-N-Verknüpfung C-C-Verknüpfung | Tyrosyl-tRNA-Synthetase 6.1.1.1 Acetyl-CoA-Synthetase 6.2.1.1 NAD-Synthetase 6.3.1.5 Pyruvatcarboxylase 6.4.1.1 |
Metalloenzyme enthalten als Bestandteil Metall-Ionen (z. B. Oxidoreductasen Fe2+ und Fe3+, Oxidasen Cu2+, Dehydrogenasen Zn2+, Nitrogenase Mo2+ und α-Amylase Ca2+) oder werden durch kooperativen Einfluß von Metall-Ionen in ihrer Wirksamkeit optimiert (z. B. Zn2+ in Acylasen und Mg2+ in Hexokinase und Carboxylase).
Die an der Bildung des aktiven Zentrums der E. beteiligten Aminosäuren sind in der Primärstruktur oftmals sehr weit voneinander entfernt, kommen aber durch die räumliche Faltung der Polypeptidkette in unmittelbare Nachbarschaft. Häufig zeigt das aktive Zentrum von E., die zur gleichen Gruppe gehören, auffallende Übereinstimmung. So enthalten z. B. die tierischen Serinproteasen Trypsin, Chymotrypsin, Elastase, Thrombin und Plasmin einen reaktiven Serinrest im aktiven Zentrum, der von Asparaginsäure- und Histidinresten umgeben ist. Für derartig verwandte E. wird eine von einem gemeinsamen Urenzym ausgehende Evolution angenommen.
Die Bildung der E. erfolgt generell nach den Prinzipien der Proteinbiosynthese. Die in der Zelle ständig gebildeten E. werden als konstitutive E. bezeichnet, die nur unter bestimmten Wachstumsbedingungen oder im Bedarfsfall produzierten E. nennt man adaptive E. Bei den letzteren unterscheidet man die induzierbaren E., die in größeren Mengen und mit erhöhter Aktivität unter dem Einfluß eines Induktors entstehen, wobei als Induktor das Substrat des betreffenden Enzyms oder Fremdmoleküle, z. B. Pharmaka oder Pflanzenschutzmittel, agieren, und die reprimierbaren E., deren Synthese durch bestimmte Stoffe, vor allem durch die Endprodukte einer Biosynthesekette, blockiert werden kann.
Wirkungsmechanismus. Bei allen Enzymreaktionen wird durch spezifische intermolekulare Wechselwirkung zwischen Enzym (E) und Substrat (S) zunächst ein Enzym-Substrat-Komplex (ES) gebildet, der durch Konformationsänderung der Proteinkomponente in einen aktivierten Komplex umgelagert wird und dann in den Enzym-Produkt-Komplex (EP) übergeht. Aus dem EP-Komplex wird durch Dissoziation das Produkt freigesetzt und das Enzym zurückgebildet. Die Teilschritte der Reaktion können wie folgt formuliert werden:
E + S ![]()
ES ![]()
EP ![]()
P + E.
Die Gleichgewichtspfeile weisen darauf hin, daß alle Reaktionsschritte reversibel verlaufen. Die Umkehrreaktionen sind besonders dann wichtig, wenn deren Umsatz an freier Energie nur gering ist, z. B. bei Umesterungen oder Transaminierungen. Durch Veränderung der Gleichgewichtskonzentrationen kann das Gleichgewicht nach beiden Seiten verschoben werden. Eine Gleichgewichtsverschiebung findet z. B. auch dann statt, wenn das Reaktionsprodukt in einer Folgereaktion schneller umgesetzt wird, als es in der ersten entsteht. Bei E., die nur in Verbindung mit einem Coenzym (C) wirksam sind, übernimmt dieses häufig einen vom Substrat (S1x) abgespaltenen Molekülteil (x), z. B. Wasserstoffatome bei Oxidoreductasen, wird dann selbst als Coenzym (Cx), z. B. CH2, von einem Enzym (E2) übernommen, das dann x, z. B. 2 H, auf ein Zweitsubstrat überträgt:
E1 + S1H2 + C ![]()
[E1C·S1H2] ![]()
E1 + S1 + CH2;
CH2 + E2 + S2![]()
[E2 ·S2 ·CH2] ![]()
E2 + C + S2H2.
Die Geschwindigkeit der enzymatisch katalysierten Reaktion ist insbesondere von einer hohen Substratkonzentration im Bereich des aktiven Zentrums, von einer optimalen Orbitalorientierung der reagierenden Moleküle sowie von der schnellen Konformationsänderung der Proteinkomponente und der Zerfallsgeschwindigkeit des EP-Komplexes abhängig. Bei gegebener Enzymmenge steigt die Reaktionsgeschwindigkeit mit steigender Substratkonzentration an. Nach Michaelis und Menten gilt für die Enzymreaktion mit einem Substrat
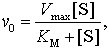
wobei v0 die Anfangsgeschwindigkeit, Vmax die maximale Geschwindigkeit, [S] die Substratkonzentration und KM die Michaelis-Menten-Konstante bedeuten. KM ist die Substratkonzentration, bei der die halbe maximale Reaktionsgeschwindigkeit erreicht wird. Hohe KM-Werte deuten darauf hin, daß die E. nur eine geringe Affinität zum Substrat haben. Im Substrat-Geschwindigkeits-Diagramm ergibt sich bei den durch die Michaelis-Menten-Gleichung charakterisierten E. ein hyperbelförmiger Verlauf der Enzymkennlinie. Allosterische E. zeigen einen sigmoiden (S-förmigen) Verlauf der Kennlinie. Hier führt die Effektorbindung zu einer sehr schnellen Änderung der dreidimensionalen Proteinstruktur, wobei die von einer Untereinheit ausgehenden Konformationsänderungen auf weitere Untereinheiten des Enzymmoleküls übertragen werden können.
Mechanistisch kann die Enzymreaktion eine Kovalenzkatalyse oder eine allgemeine Säure-Base-Katalyse sein. Bei der Kovalenzkatalyse wird ein hochreaktives Intermediat aus Enzym und Substrat gebildet, z. B. Acylverbindungen der Serinenzyme oder Schiffsche Basen der Lysinenzyme (Transaldolase, D-Aminosäureoxidase). Die Katalyse wird durch den Angriff einer nucleophilen Gruppe des E. (Imidazol des Histidins, OH-Gruppe des Serins, SH-Gruppe des Cysteins) auf ein elektrophiles C-Atom des Substrats eingeleitet. Ein Beispiel für die Kovalenzkatalyse ist die Spaltung spezifischer Peptidbindungen durch das Serinenzym Chymotrypsin (Abb.). Im aktiven Zentrum des Enzyms sind die Aminosäurereste Serin195, Histidin57 und Asparaginsäure102 an der Katalyse beteiligt. Das Asparaginsäureanion übernimmt die Funktion einer Cobase, das Histidin die der allgemeinen Base und das Serin die des Nucleophils. Das beim nucleophilen Angriff des Serins auf die Carbonylgruppe der Peptidbindung intermediär gebildete Acylenzym wird unter Mitwirkung von Wasser hydrolysiert. Bei der allgemeinen Säure-Base-Katalyse ermöglichen E. chem. Reaktionen im neutralen pH-Bereich. Es werden solche Reaktionen beschleunigt, die ohne Katalysator hohe OH-- oder H+-Konzentrationen erfordern würden. Die Katalyse wird hier durch Protonendonor oder Protonenakzeptorgruppen des aktiven Zentrums bewirkt, z. B. durch die COOH- (COO--) Gruppe von Asparaginsäure, die NH3+- (-NH2-) Gruppe von Lysin oder die -C6H5OH- (-C6H5O--) Gruppe des Tyrosins.
Enzymhemmung. Generell kann die Wirkung von E. durch Effektoren beeinflußt werden. Aktivatoren erhöhen die Enzymaktivität z. B. durch spezifische Bindung inaktivierender Metallionen oder durch Erhöhung der Zerfallsgeschwindigkeit des Enzym-Substrat-Komplexes. Inhibitoren bewirken eine im allgemeinen reversible Hemmung, wobei man eine kompetitive und eine nichtkompetitive Hemmung unterscheidet. Zu einer kompetitiven Hemmung kommt es, wenn natürliches Substrat und diesem in der Struktur ähnliche Verbindungen (Inhibitoren, Antimetabolite) um dieselbe Bindungsstelle am Enzym konkurrieren und das betreffende E. keine absolute Substratspezifität aufweist. So wird z. B. Succinatdehydrogenase, die die Umsetzung von Bernsteinsäure als natürliches Substrat katalysiert, durch die um eine CH2-Gruppe ärmere Malonsäure gehemmt. Die kompetitive Hemmung kann grundsätzlich durch Erhöhung der Substratkonzentration zurückgedrängt oder aufgehoben werden. Bei der nicht-kompetitiven Hemmung ist eine strukturelle Verwandtschaft des Inhibitors zum Substrat nicht erforderlich. Die hemmende Wirkung ist von der Inhibitorkonzentration abhängig und beruht auf der reversiblen Wechselwirkung des Inhibitormoleküls mit funktionellen Enzymgruppen, die für die Ausbildung der katalytisch wirksamen Proteinkonformation verantwortlich sind. So werden z. B. E., die eine essentielle SH-Gruppe enthalten, nichtkompetitiv durch Schwermetall-Ionen gehemmt. Andererseits wirken Chelatbildner, z. B. EDTA, als nichtkompetitive Inhibitoren, indem sie für die Enzymwirkung essentielle Metall-Ionen binden. Natürliche Inhibitoren spielen eine wichtige Rolle für die Regulation der Aktivität von E. in der lebenden Zelle.
;)
Enzyme. Abb.: Vereinfachte Darstellung der Spaltung einer Peptidbindung durch Chymotrypsin. R1 und R2 bedeuten Seitenketten der Aminosäuren 1 und 2.
Enzymeinheiten. Zur Bestimmung der Enzymaktivität wird in der durch eine bestimmte Enzymmenge katalysierten Reaktion die zeitliche Abnahme des Substrates oder die Zunahme des Substrates oder die Zunahme des Reaktionsproduktes meist spektroskopisch ermittelt.
Nach Festlegungen der Internationalen Enzymkommission der IUPAC ist eine Enzymeinheit (1 U) die Menge an E., die unter Standardbedingungen die Umwandlung von 1 μmol Substrat pro Minute katalysiert. Als neue Internationale Einheit wurde 1972 die katalytische Einheit Katal, Einheitenzeichen kat, eingeführt. 1 kat ist die Menge an Enzymaktivität, die 1 mol Substrat pro Sekunde umsetzt. Als Untereinheiten wurden das Mikrokatal (μkat), Nanokatal (nkat) und Picokatal (pkat) zugelassen. Für die Umrechnung zwischen den Einheiten gilt: 1 kat = 6·107 U bzw. 1 U = 16,67 nkat.
Die durch ein Enzymmolekül oder durch ein aktives Zentrum je Minute umgesetzte Anzahl an Substratmolekülen wird als molekulare Aktivität (früher Wechselzahl) bezeichnet. Mit dem Umsatz von 36 Millionen Substratmolekülen je Minute zeigt das E. Carboanhydrase C eine besonders hohe Aktivität.
Gewinnung. E. können aus tierischen, pflanzlichen oder mikrobiellen Vorkommen gewonnen werden. Zur Isolierung aus tierischem Gewebe werden meist nur bestimmte Organe, z. B. Bauchspeicheldrüsen oder Nieren, aufgearbeitet. Nach Homogenisierung des Materials werden die E. direkt mit geeigneten Pufferlösungen extrahiert oder zunächst bei tiefen Temperaturen mit einem organischen Lösungsmittel, z. B. Aceton, zu einem Trockenpulver verarbeitet. Pflanzliche E., z. B. Papain, werden aus den Preßsäften isoliert, die man aus dem mechanisch zerkleinerten Pflanzenmaterial erhält. Die Konzentrierung und Feinreinigung der E. erfolgt durch Fällungs- und Adsorptionsverfahren sowie durch Ultrafiltration.
Für die technische Gewinnung von E. hat die Fermentation mit Mikroorganismen und Mutanten überragende Bedeutung erlangt. Die Produktion erfolgt diskontinuierlich in Fermentern bis 100000 l Inhalt, die Fermentationsdauer beträgt 50 bis 150 h. Die Isolierung ist einfach, wenn die E. extrazellulär in das Kulturfiltrat ausgeschieden werden, z. B. bei den im Maßstab von 500 t/a produzierten bakteriellen Proteasen und Amylasen. Die meisten E. werden intrazellulär gebildet, so daß zunächst die meist stabile Zellwand der Mikroorganismen mechanisch zerstört werden muß, z. B. im Hochdruckhomogenisator oder in Rührwerkskugelmühlen.
Bedeutung und Verwendung. Die Vorzüge der Enzymkatalyse, unter milden Bedingungen nebenproduktfreie Reaktionen mit hoher Ausbeute zu ermöglichen, werden in zunehmendem Maße in der Praxis genutzt. Technische Einsatzgebiete sind vor allem die Waschmittel-, Lebensmittel-, Getränke- und Pharmaindustrie (Tab. 2). Ein bedeutender Fortschritt in der Enzymtechnik wurde durch die Immobilisierung von E. erreicht (immobilisierte Enzyme).
Enzyme. Tab. 2: Technische Verwendung von Enzymen (Auswahl).
| |||
| Proteasen (Trypsin, Elastase, Pepsin, Papain, mikrobielle Enzyme u. a.) | Hydrolyse von Proteinen | Waschmittelzusatz (0,01 ... 0,025 %), hydrolytischer Abbau eiweißhaltiger Verschmutzungen; Hydrolyse von Sojabohnenprotein (Sojasoßenherstellung), Backhilfsmittel (Partialhydrolyse von Kleber), Proteinhydrolysatherstellung aus Fleischabfällen und Fischproteinen | |
| α-Amylase (Pankreas, Bacillus sp.) | Hydrolyse von Stärke zu Dextrinen und Maltose | Verdauungs- und Backhilfe, Stärkekleisterherstellung, Stärkeentfernung in der Textilindustrie Entschlichtung) | |
| Glucoamylase (Aspergillus sp.) | Exohydrolyse von Stärke | Stärkeabbau zu Glucose in hoher Ausbeute und Reinheit | |
| Glucoseisomerase (Streptomyces sp., Bacillus sp.) | Isomerisierung von D-Glucose und D-Fructose | Herstellung von Flüssigzucker hoher Süßkraft ("Fructosesirup") | |
| Glucoseoxidase (Aspergillus sp., Penicillium sp.) | Oxidation von Glucose zu Gluconolacton | Konservierung von Nahrungsmitteln und Getränken durch O2-Entfernung | |
| Invertase (Saccharomyces sp. und Aspergillus sp.) | Spaltung von Saccharose in Glucose und Fructose | Herstellung von Invertzucker mit hoher Süßkraft | |
| Katalase (Pankreas oder Rhizopus nigricans) | Zersetung von H2O2 | in Kombination mit Glucoseoxidase zur Konservierung von Nahrungsmitteln | |
| Lipase (Pankreas, Aspergillus sp.) | Fettspaltung | Gewinnung empfindlicher Fettsäuren, Beschleunigung der Käsereifung | |
| Cellulase (Aspergillus niger und Stachybotysatra) | Hydrolyse von Cellulose zu Cellobiose | Aufschluß von Nahrungsmitteln, Glucosegewinnung aus cellulsehaltigen Rohstoffen | |
| Lactase (Aspergillus sp.) | Spaltung von Lactose in Galactose und Glucose | Herstellung von Diätmilch | |
| Pektinesterase | Hydrolyse von Polygalacturonsäure | Beseitigung von Trübungen in Fruchtsäften und Bier, Entfernung von Pektinhüllen von pflanzlichen Fasern | |
| Penicillinamidase (Bacillus sp.) | Bildung von 6-Aminopenicillansäure aus Penicillin G | Herstellung halbsynthetischer Penicilline | |
| L-Aminosäureacylasen (Niere, Aspergillus oryzae) | enantioselective Hydrolyse von DL-Acylaminosäuren in L-Aminosäuren und D-Acylaminosäuren | Produktion essentieller Aminosäuren für die tierische und menschliche Ernährung | |
| Streptokinase (Streptokokken) | Plasminogen → Plasmin | Beseitigung von Blutgerinseln (Fibrinolyse) |
Eine Alternative zu diesen trägerfixierten E. ist der Einsatz von Enzym-Membran-Reaktoren (Membranreaktor) in der Biotechnologie.
In der Medizin dienen Bestimmungen der E. im Serum oder im Harn zur differentiellen Diagnose und Therapiekontrolle zahlreicher Erkrankungen, z. B. Herzinfarkt, Hepatitis, Pankreatitis. Bestimmte E. werden zur Substitutionstherapie bei Störungen der Verdauung, Blutgerinnung und Fibrinolyse sowie zur Behandlung von Verbrennungen, Wunden, Transplantaten, Herz-, Kreislauf- und Krebserkrankungen zugeführt.
Von Bedeutung sind Enzymimmunoassays, die E. als "Marker" für immunologische Reaktionspartner verwenden und eine schnelle Konzentrationsbestimmung von Hormonen, Immunoglobulinen, Antigenen, Drogen u. a. ermöglichen. Enzymelektroden dienen zur Erfassung von Meßgrößen elektrochem. Prozeßkontrollen. Am bekanntesten sind Glucose-sensitive Elektroden mit immobilisierter Glucoseoxidase als E. Gemessen wird hier die Abnahme der Sauerstoff- oder die Zunahme der Wasserstoffperoxidkonzentration. Durch Kopplung der Glucoseoxidase mit entsprechenden Hydrolasen erhält man Enzymelektroden, die zur schnellen Bestimmung glucosehaltiger Oligosaccharide verwendet werden. Enzymthermistoren nutzen die bei enzymkatalysierten chem. Umsetzungen auftretende Reaktionswärme als Meßsignal.
Die Anwendung von E. und gentechnisch hergestellten Mutanten wird in Zukunft sowohl auf dem Sektor der Analytik als auch auf dem Gebiet der Enzymtechnik (Biotransformationen) an Umfang zunehmen. Das gilt auch für den Einsatz von katalytischen Antikörpern sowie auch von künstlichen, proteinfreien E., den Synzymen, bei denen eine zum Teil beträchtliche katalytische Aktivität durch den Einbau "enzymspezifischer" Strukturelemente in synthetische Makromoleküle erreicht wird.







Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.