Lexikon der Chemie: Massenwirkungsgesetz
Massenwirkungsgesetz, Abk. MWG, Gesetz über die Gleichgewichtskonzentrationen einer chem. Reaktion, das lautet: Das Produkt aus den Konzentrationen der Endstoffe, dividiert durch das Produkt der Konzentrationen der Ausgangsstoffe im chem. Gleichgewicht ist eine Konstante, die Gleichgewichts- oder Massenwirkungskonstante Kc. Dabei treten die stöchiometrischen Koeffizienten als Exponenten der zugehörigen Konzentrationen auf. Das M. gilt in dieser Form nur für ideale Mischungen. Treten zwischen den Teilchen Wechselwirkungen auf, ist Kc nicht mehr konstant, sondern verändert sich mit den Anfangskonzentrationen. Deshalb werden anstelle der Konzentrationen die Aktivitätenai eingeführt.
Für die allgemeine chem. Reaktion
lautet das M. in strenger Form
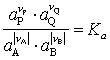
.
Dabei sind νi stöchiometrische Faktoren, die als Exponenten der zugehörigen Aktivitäten auftreten. Die νi der Ausgangsstoffe sind nach der thermodynamischen Vorzeichenkonvention negativ, gehen aber im M. positiv ein (|νA|,|νB|). Die Massenwirkungskonstante ist für jede chem. Reaktion eine charakteristische Konstante, die nicht von den Mengen und Mischungsverhältnissen der Reaktanten abhängt, sich aber mit der Temperatur und dem Druck ändert. Beteiligen sich bei heterogenen Reaktionen kondensierte Phasen reiner Stoffe am Gleichgewicht, ist zu beachten, daß die Aktivitäten reiner Stoffe definitionsgemäß gleich eins sind, d. h., sie treten im M. nicht auf. So gilt z. B. für das Boudouard-Gleichgewicht CO2 + C ![]()
2 CO; Ka = a2CO/aCO2, da ac = 1.
Wendet man das M. auf Reaktionen in idealen Mischungen an, so sind in den Aktivitäten ai = fixi die Aktivitätskoeffizienten fi = 1, und das M. kann mit den Molenbrüchen xi formuliert werden. Zur Kennzeichnung schreibt man für die Massenwirkungskonstante dann das Symbol Kx. Außerdem ist eine Umrechnung der Molenbrüche auf andere Zusammensetzungsgrößen möglich, z. B. auf Konzentrationen ci, Molalitäten mi, Partialdrücke pi bei Gasreaktionen oder Stoffmengen ni. Dabei ändern sich die Zahlenwerte und Einheiten der Massenwirkungskonstanten, die zur Unterscheidung mit dem jeweiligen Index (c, m, p, n) versehen werden. Es gelten folgende Zusammenhänge: a) für ideale Systeme Ka = Kx, b) für Gasreaktionen in idealen Mischungen Kx = Kppi-Σνi = Kc(RT/p)Σνi, c) für ideal verd. Lösungen Kx= Kc(M1/ρ1) iΣνi = Km(M1)Σνi. Dabei sind Σνi = νP + νQ – |νA| – |νB|, p der Gesamtdruck, R die Gaskonstante, T die absolute Temperatur, M1 die Molmasse und ρ1 die Dichte des Lösungsmittels. Für Reaktionen ohne Änderung der Molzahl, d. h. Σνi = 0, sind alle K-Werte gleich und ohne Einheit.
Das M. läßt sich thermodynamisch aus der van't-Hoffschen Reaktionsisotherme ΔRG = ΔRG0 + RT Σνiln ai ableiten (Thermodynamik, 2. Hauptsatz, Anwendung d). Im Gleichgewicht ist die freie Reaktionsarbeit ΔRG = 0, und es folgt Ka = e-ΔRG0/RT, wobei ΔRG0 die freie Reaktionsarbeit unter Standardbedingungen bezeichnet. Die letzte Beziehung bildet den Ausgangspunkt zur thermodynamischen Berechnung von Gleichgewichtskonstanten (Ulichsche Näherungen). Für die Temperaturabhängigkeit der Gleichgewichtskonstanten bei konstantem Druck folgt die van't-Hoffsche Reaktionsisobare.
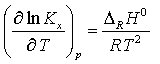
bei konstantem Volumen die van't-Hoffsche Reaktionsisochore
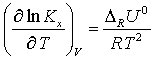
und für die Druckabhängigkeit von Kx bei konstanter Temperatur
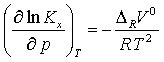
mit ΔRH0 = molare Reaktionsenthalpie, ΔRU0 = molare Reaktionsenergie und ΔRV0 = Änderung des Volumens der Mischung bei 1 mol Formelumsätzen (Le-Chateliersches Prinzip) jeweils unter Standardbedingungen.
Im Falle der formulierten Gleichgewichtsreaktion gilt für die Hinreaktion das kinetische Zeitgesetz r1 = k1cA|νA| cB|νB| und für die Rückreaktion r-1 = k-1cPνPcQνQ, wobei r Reaktionsgeschwindigkeit und k Geschwindigkeitskonstante (Reaktionskinetik) bedeuten. Voraussetzung für die Formulierung der Zeitgesetze ist, daß beide Teilschritte Elementarreaktionen sind. Mit Hilfe des Prinzips der mikroskopischen Reversibilität (Gleichgewicht) läßt sich jedoch zeigen, daß diese Art der Ableitung auch auf komplexe Reaktionen übertragbar ist. Während der Einstellphase des Gleichgewichtes ist die effektiv meßbare Geschwindigkeit r = r1 – r-1. Im Gleichgewicht gilt r = 0. Hin- und Rückreaktionen verlaufen gleich schnell, makroskopisch findet keine Konzentrationsänderung mehr statt. Es liegt ein dynamisches Gleichgewicht vor. Es folgt
k1cA|νA| cB|νB| = k-1cPνPcQνQ oder
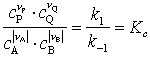
.
d. h. das M. mit der Gleichgewichtskonstanten Kc. Berücksichtigt man, daß für beide Geschwindigkeitskonstanten die Eyring-Gleichung gilt, so erhält man Kc = K1/K-1 = e-(ΔG1≠ – ΔG-≠1)/RT= e-(ΔS1≠ – ΔS-≠1)/R = e-(ΔH1≠ – ΔH-≠1)/RT wobei ΔG≠, ΔS≠ und ΔH≠ die freien Aktivierungsenthalpien, die Aktivierungsentropien und die Aktivierungsenthalpien von Hin- (1) und Rückreaktion (-1) bezeichnen. Durch Vergleich mit der Beziehung Kx= e-ΔRG0/RT ergeben sich die folgenden Zusammenhänge zwischen den thermodynamischen Reaktionsgrößen und den kinetischen Aktivierungsparametern: ΔRG0 = ΔG1≠ – ΔG≠-1, ΔRS0 = ΔS1≠ – ΔS≠-1, und ΔRH0 = ΔH1≠ – ΔH≠-1.
Die Aussagen des M. können dazu dienen, die Ausbeute einer Reaktion gezielt zu beeinflussen, und zwar a) über die Konzentrationen (Aktivitäten) der Ausgangsstoffe bei unveränderten äußeren Bedingungen (K = konst.) und b) durch Veränderung von K aufgrund von Temperatur- und/oder Druckänderungen. a) Erhöht man die Konzentration eines Ausgangsstoffes, erfolgt so lange eine Reaktion in Richtung der Produkte, bis der Wert von K wieder erreicht ist, d. h., die Ausbeute steigt. Die wertvolleren Reaktanten können durch einen Überschuß der billigeren besser ausgenutzt werden. Den gleichen Effekt hat die ständige Entfernung von Produkten aus der Reaktionsmischung. b) Durch Veränderung von Temperatur oder Dr uck kann K vergrößert oder verkleinert und damit das Gleichgewicht in Richtung Produkte oder Ausgangsstoffe verschoben werden (Le-Chateliersches Prinzip).
Zur Anwendung des M. auf Elektrolytlösungen z. B. pH-Wert, Dissoziationskonstanten (Dissoziation) von Säuren und Basen, Ostwaldsches Verdünnungsgesetz, Löslichkeitsprodukt, Hydrolyse, Pufferlösung. Das M. bildet die Grundlage analytischer Meßmethoden und ermöglicht die Berechnung von Ausbeuten und deren gezielte Steuerung bei technischen Synthesen.







Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.