Lexikon der Chemie: Röntgenspektroskopie
Röntgenspektroskopie, Röntgenspektralanalyse, Teilgebiet der Spektroskopie, das sich mit der Wechselwirkung von Röntgenstrahlung (elektromagnetisches Spektrum) und materiellen Systemen beschäftigt. Die R. dient hauptsächlich zum qualitativen und quantitativen Nachweis chem. Elemente. Man unterscheidet Röntgenabsorptionsspektroskopie, Röntgenemissionsspektroskopie und Röntgenfluoreszenzanalyse. Die Röntgenabsorptionsspektroskopie mißt die beim Durchgang von Röntgenstrahlung durch eine Probe auftretende Intensitätsabnahme. Die Röntgenemissionsspektroskopie und die Röntgenfluoreszenzanalyse untersuchen die von einer entsprechend angeregten Probe emittierte Röntgenstrahlung, wobei die Anregung im ersten Fall meist durch Elektronenstoß, im zweiten Fall durch Photonen entsprechender Energie erfolgt.
Bremsstrahlung und Eigenstrahlung. Röntgenstrahlen entstehen, wenn Elektronen hoher Geschwindigkeit etwa durch Auftreffen auf feste Stoffe (z. B. die Anode der Röntgenröhre) plötzlich abgebremst werden. Die maximale Energie Emax des von einem Elektron ausgelösten Röntgenstrahls entspricht der vollständigen Umwandlung der kinetischen Energie des Elektrons in die Energie des Strahlungsquants.
,
wobei e die Ladung des Elektrons, U die angelegte Spannung, h das Plancksche Wirkungsquantum, ν die Frequenz, λ die Wellenlänge der Strahlung und c die Lichtgeschwindigkeit bedeuten. Durch einfache Umformung erhält man daraus das Duane-Huntsche Gesetz
,
das die kurzwellige Grenze der entstehenden Röntgenstrahlung, die durch die Spannung U der Röhre vorgegeben und vom Anodenmaterial in erster Näherung unabhängig ist, angibt. Da im allg. die Elektronen nicht ihre gesamte Energie in einem Bremsakt abgeben, ist für den größten Teil der ausgesandten Röntgenstrahlung die Wellenlänge größer als durch das Duane-Huntsche Gesetz angegeben. Die Bremsstrahlung besteht aus einem Kontinuum, das eine scharfe kurzwellige Grenze bei λmin hat (Abb. 1). Läßt man durch allmähliche Steigerung der Spannung die kurzwellige Grenze sich mehr und mehr in das Gebiet kurzer Wellenlängen verschieben, so beobachtet man je nach dem Anodenmaterial an bestimmten Stellen im Spektrum das Auftreten einer sehr starken Strahlung (Abb. 2). Man bezeichnet diese Strahlung als charakteristische Röntgenstrahlung oder Eigenstrahlung, d. h., daß die Wellenlänge dieser Strahlung durch das Material der Anode bestimmt wird. Diese Eigenstrahlung bildet die Grundlage der Röntgenemissionsspektroskopie und Röntgenfluoreszenzanalyse.
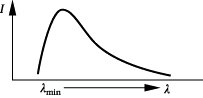
Röntgenspektroskopie. Abb. 1: Bremsspektrum.
;)
Röntgenspektroskopie. Abb. 2: Überlagerung von Bremsstrahlung und Eigenstrahlung.
Entstehung der Röntgenspektren. Führt man Atomen entsprechend große Energiebeträge zu, indem man z. B. Elektronen hoher Energie auf sie auftreffen läßt, so können Elektronen aus den inneren Bahnen der Atome herausgeschleudert werden. Die dabei in den inneren Schalen entstandenen Lücken werden wieder aufgefüllt, indem Elektronen aus weiter außen liegenden Schalen in diese Lücke hineinspringen und die dabei frei werdende Energie in Form von Röntgenstrahlen entsprechend der Gleichung E2 – E1 = h·ν emittieren.
;)
Röntgenspektroskopie. Abb. 3: Entstehung der Röntgenspektren.
Die Wellenlängen dieser charakteristischen Röntgenstrahlung sind durch die Abstände der inneren Energieniveaus des Atoms bestimmt und damit für die betreffende Atomsorte charakteristisch. Wird ein Elektron aus der innersten oder K-Schale herausgeschlagen, so beobachtet man die K-Serie, die durch Elektronenübergänge aus der L-Schale, M-Schale usw. in die K-Schale zustande kommt. Wie Abb. 3 zeigt, werden die entsprechenden Linien als Kα-, Kβ-, Kγ-Linien bezeichnet. Wird bei der Anregung ein Elektron aus der L-Schale entfernt, so entsteht durch Elektronenübergänge aus höheren Schalen die L-Serie. Die meisten Elemente haben mehrere Serien, die in verschiedenen Spektralbereichen liegen. Die verschiedenen Serien unterscheiden sich durch ihre Linienanordnung, dagegen hat die gleiche Serie bei verschiedenen Elementen eine analoge Struktur. Für viele Elemente liegen ein oder zwei Serien vor, nämlich für die leichteren Elemente die K-Serie, für die etwas schwereren dazu die L-Serie. Bei Atomen mit hohen relativen Atommassen können noch M-, N-, O- und P-Serien hinzukommen. Mit dem Auftreten der K-Serie werden Elektronenlücken in der L-, M-, N-Schale frei, zu deren Auffüllung erneut ein Elektron aus einer äußeren Schale einspringt. Mit dem Entstehen der Kα-Linie ist also außer dem Auftreten der übrigen Linien der K-Serie notwendigerweise auch das Auftreten der L-, M- und N-Serien – je nach Ordnungszahl des Elements – verknüpft. Die Existenz des Röntgenbremskontinuums bedingt, daß Röntgenspektren bei Elektronenanregung stets aus einem kontinuierlichen Untergrund mit überlagerten Linien der charakteristischen Strahlung bestehen. Die Frequenz der Linien im Röntgenspektrum wird durch das MoseleyscheGesetz bestimmt. Danach wächst für analoge Linien verschiedener Elemente (z. B. die Kα-Linie) die Frequenz proportional mit dem Quadrat der um eine Konstante verminderten Ordnungszahl: ν ≈ (Z – a)2, wobei ν die Frequenz der Eigenstrahlung eines Elementes mit der Ordnungszahl Z und a die Abschirmungskonstante bedeuten, die im Falle der Kα-Strahlung 1 ist, da durch das zweite Elektron der K-Schale eine Kernladung abgeschirmt wird.
Genauere Untersuchungen haben gezeigt, daß die in Abb. 3 dargestellten Übergänge weiter aufgespalten sind. Dies rührt daher, daß jeder Energiezustand der Hauptquantenzahl n in Wirklichkeit aus einer Gruppe von 2n – 1 mehr oder weniger eng benachbarten Zuständen besteht. Die K-Schale besteht nur aus einem einzigen Zustand, die L-Schale aus 3 (mit LI, LII, LIII bezeichnet), die M-Schale aus 5, so daß z. B. verschiedene Übergänge zwischen L- und K-Schale auftreten können. Allerdings treten nicht alle diese theoretisch möglichen Übergänge tatsächlich auf, da für sie bestimmte Auswahlregeln gelten. Weiterhin ist zu beachten, daß auch strahlungslose Übergänge zwischen inneren Niveaus erfolgen können, wobei ein weiteres Elektron emittiert wird (Auger-Elektronen-Spektroskopie). Durch diesen Effekt wird die Strahlungsausbeute insbesondere bei leichten Elementen gemindert.
Apparatives. Der Aufbau eines Röntgenspektrometers gleicht prinzipiell dem anderer Spektrometer (Spektralapparaturen). Man unterscheidet 3 Hauptbauelemente: 1) Anregungsquelle, 2) Monochromator, 3) Detektor. Bei Röntgenemissionsspektren unterscheidet man Primärspektren, die meist durch schnelle Elektronen, aber auch durch Protonen oder Ionen angeregt werden, und Sekundärspektren, die man durch Anregung mit Röntgenstrahlen erzeugt. Um das Röntgenspektrum einer Substanz bei primärer Anregung mit Elektronen zu messen, bringt man die Probe auf die Anode einer zerlegbaren Röntgenröhre auf. Der Nachteil dieser Methode liegt in der starken Erhitzung der Probe und ihrer möglichen chem. Umwandlung. Schonender ist die Aufnahme von Sekundärspektren, wo meist mit dem Bremsspektrum einer Wolframanode, aber auch mit Kα- oder Lα-Strahlung angeregt wird. Wegen des fehlenden Bremsuntergrundes sind Sekundärspektren meist kontrastreicher als Primärspektren. Das zur Aufnahme von Absorptionsspektren erforderliche intensive kontinuierliche Röntgenspektrum wird gewöhnlich aus einer Röntgenröhre mit Wolframanode bezogen.
Die spektrale Zerlegung der Röntgenstrahlung im Monochromator erfolgt durch Einkristalle oder geritzte Gitter entsprechend der durch das Braggsche Gesetz gegebenen Beziehung. Für Untersuchungen im Bereich λ < 2 nm werden meist Kristalle, für längere Wellenlängen (λ > 2 nm) Gitter eingesetzt. Durch Drehung des Analysatorkristalls oder des Gitters und der damit verbundenen Variation des Glanzwinkels läßt sich monochromatische Strahlung erhalten. Diese Verfahren der spektralen Zerlegung von Röntgenstrahlen werden als wellenlängendispersive Verfahren bezeichnet. Daneben sind noch energiedispersive Verfahren in Anwendung. Die Strahlung gelangt hier, ohne daß vorher ein Monochromator eingeschaltet wurde, direkt in den Detektor (speziell Halbleiterdetektor). In ihm werden nicht nur die Röntgenquanten angezeigt, sondern auch ihre Energie bestimmt. Der Detektor übernimmt hier gleichzeitig die Aufgabe des dispergierenden Elements. Die energiedispersive Strahlungszerlegung weist ein geringeres Auflösungsvermögen als die wellenlängendispersive auf und scheidet für die Untersuchung der Feinstruktur von Spektren aus.
Als Detektoren werden bei den wellenlängendispersiven Methoden in zunehmendem Maße Quantendetektoren verwendet. In beiden Fällen ist die Anpassung an den zu vermessenden Spektralbereich erforderlich. Als Quantendetektoren werden z. B. Ionisations- und Szintillationszähler, Sekundärelektronenvervielfacher oder Halbleiterzähler eingesetzt. Der spezielle Aufbau des Spektrometers wird durch den Wellenlängenbereich, für den es vorgesehen ist, bestimmt. Man unterscheidet offene oder Kurzwellenspektrometer (λ < 0,2 nm), Vakuum- oder Langwellenspektrometer (λ ≈ 0,2 bis 1 nm) und Hochvakuum- oder Ultralangwellenspektrometer (λ > 2 nm).
Röntgenemissionsspektren. Sie werden erhalten, wenn die bei der Anregung einer Probe mit Elektronen oder Röntgenstrahlen emittierte Strahlung spektral zerlegt wird. Die Röntgenemissionsspektren beinhalten die für die Elementanalyse notwendigen Informationen. Bei der qualitativen Analyse bestimmt man die Wellenlänge der von der Probe emittierten Spektrallinien und ordnet sie mit Hilfe von Tabellen den Elementen zu. Der sichere Nachweis eines Elementes ist dann erbracht, wenn mehrere Linien einer Spektralserie dieses Elementes identifiziert worden sind. Für die quantitative Analyse werden zusätzlich die Intensitäten der Spektrallinien benötigt. Gewöhnlich verwendet man zur Auswertung die intensitätsstarken Kα- oder Lα-Linien. Die Auswertung geschieht mit Hilfe von Eichproben. Aus der Wellenlänge der Röntgenhauptlinien Kα, Lα usw. kann man die Energiedifferenz zwischen den inneren Elektronenniveaus entnehmen (Abb. 3) und aus der Wellenlänge der kurzwelligsten Linien einer Serie die Energiedifferenz zwischen innerem Niveau und optischem Niveau. Diese Differenzen hängen vom Bindungszustand der emittierenden Atome ab, so daß die Anwendung der Röntgenemissionsspektren in der Strukturanalyse möglich ist.
Spezielle, für die Praxis wichtige Verfahren der Röntgenemissionsspektroskopie sind die Methoden der Röntgenfluoreszenzanalyse sowie der Elektronenstrahlmikroanalyse.
Röntgenabsorptionsspektren. Sie werden vermessen, indem man die Schwächung eines Röntgenstrahlbündels beim Durchgang durch eine Schicht eines Stoffes in Abhängigkeit von der Wellenlänge registriert. Die Absorption folgt dem Lambert-Beerschen-Gesetz. Die Stärke der Absorption wird durch die Art des absorbierenden Materials, d.h. seinen Massenschwächungskoeffizienten (MSK) bestimmt: MSK = μ/ρ, wobei μ der lineare Schwächungskoeffizient und ρ die Dichte ist. Vernachlässigt man die beim Durchgang von Röntgenstrahlen durch Materie auftretenden Streuprozesse, so werden durch die absorbierten Röntgenstrahlen Elektronen aus der Probe herausgeschlagen bzw. in einen hochangeregten Zustand überführt. Eine Absorption der Kα- oder Kβ-Linie ist offensichtlich nicht möglich, da die L- und M-Schale vollbesetzt ist, so daß die K-Elektronen nicht in sie hineinspringen können. Durch Absorption kann ein K-Elektron nur in eines der unbesetzten optischen Niveaus oder in den kontinuierlichen Bereich oberhalb der Ionisierungsgrenze übergehen. Praktisch bedeutet dies, daß ein K-Elektron nur das Seriengrenzkontinuum der K-Serie absorbieren kann. Es erstreckt sich von der Ionisierungsgrenze des betreffenden Niveaus mit abnehmender Intensität nach kurzen Wellenlängen hin. Röntgenabsorptionsspektren bestehen deshalb nicht aus Linien. Trägt man die Absorption bzw. den MSK in Abhängigkeit von der Wellenlänge auf (Abb. 4), so erkennt man, daß der Massenschwächungskoeffizient bei Erreichung dieser Ionisierungsgrenze steil ansteigt. Man bezeichnet diese sprunghafte Zunahme des Absorptionskoeffizienten als K-Absorptionskante. Röntgenquanten mit dieser oder einer höheren Energie können Elektronen aus der K-Schale herausschlagen, Röntgenquanten niederer Energie jedoch nicht.
;)
Röntgenspektroskopie. Abb. 4: Abhängigkeit des Massenschwächungskoeffizienten (MSK) von der Wellenlänge.
Daß der Absorptionskoeffizient an der langwelligen Seite der Absorptionskante nicht auf 0 fällt, liegt daran, daß hier noch die Ausläufer des Seriengrenzkontinuums der L-Schale wirksam werden. Die Lage der Absorptionskanten ist charakteristisch für jedes Element und kann zu dessen Nachweis dienen (qualitative Analyse). Aus der Höhe der Absorptionskante können quantitative Aussagen erhalten werden. Bei höherer Auflösung findet man häufig eine Feinstruktur der Absorptionskanten (XANES, Abk. für engl. x-ray absorptions near edge structure), die z. B. auf die bereits erwähnte Aufspaltung der L-, M- und N-Niveaus in Unterniveaus bzw. die Möglichkeit der Überführung eines Elektrons in eines der unbesetzten optischen Niveaus zurückgeht. Weiter hängt die genaue Lage der Absorptionskanten vom Bindungszustand des betreffenden Atoms ab.
Von den röntgenspektroskopischen Methoden finden die emissionsspektroskopischen Verfahren eine breitere Anwendung als die absorptionsspektroskopischen. Neben mehr theoretischen Aussagen zum Atombau und zum Bindungsverhalten liefert die R. Aussagen zum qualitativen und vor allem quantitativen Nachweis von Elementen auch im Spurenbereich. Sie spielt deshalb in der chem. Analytik, in der Umweltanalytik, in der Kriminalistik und ähnlichen Gebieten eine wichtige Rolle. Aufgrund ihrer vielseitigen Anwendungsmöglichkeiten, des hohen Nachweisvermögens und der großen Zuverlässigkeit werden röntgenspektroskopische Methoden zur Produktions- und Qualitätskontrolle in der chem. Industrie, Metallurgie, Silicatindustrie und in anderen Bereichen eingesetzt.








Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.