Lexikon der Chemie: Thermodynamik
Thermodynamik, Teilgebiet der Physik und der physikalischen Chemie, das sich mit Aussagen über die Eigenschaften thermodynamischer Systeme beschäftigt. Der Zustand eines solchen stofflichen Systems wird durch Sätze von Parametern (Zustandsgröße) eindeutig beschrieben. Änderungen des Zustandes sind mit einem Energieaustausch und mit der Umwandlung verschiedener Energieformen ineinander, z. B. von mechanischer oder elektrischer Arbeit in Wärme, verbunden.
I) Phänomenologische oder Gleichgewichts-T. Sie ist eine rein makroskopische Wissenschaftsdisziplin, für die es ohne Belang ist, daß Stoffe eine molekulare Struktur aufweisen. Untersucht werden stoffliche Systeme im thermodynamischen Gleichgewicht. Experimentelle Meßgrößen, wie Temperatur, Druck, Volumen, chem. Zusammensetzung und Wärmekapazitäten, dienen zur Charakterisierung der Gleichgewichtszustände. Diese Zustandsgrößen sind durch Zustandsfunktionen teilweise miteinander verknüpft. Die T. liefert quantitative Aussagen über die mit Zustandsänderungen verbundenen energetischen Änderungen (Reaktionswärme, Phasenumwandlungswärme), die Richtung und die Triebkraft von Prozessen (Affinität), die dabei maximal gewinnbare Nutzarbeit und die Lage von Gleichgewichten. Über Nichtgleichgewichtsvorgänge sind nur qualitative Aussagen möglich. Die Geschwindigkeit, mit der Gleichgewichtszustände erreicht werden, ist nicht Gegenstand thermodynamischer Untersuchungen. Für chem. Reaktionen werden Zeitabhängigkeiten in der Reaktionskinetik und in der T. irreversibler Prozesse (s. u.) untersucht. Historisch hat sich die Gleichgewichts-T. zuerst entwickelt. Grundlage sind die Hauptsätze der T., die auf Erfahrung beruhen und zur Definition der Zustandsgrößen Temperatur, innere Energie und Entropie führen.
0. Hauptsatz der T.: Sind zwei Körper mit einem dritten im thermischen Gleichgewicht, so sind sie auch untereinander im thermischen Gleichgewicht. Daraus folgt, daß für makroskopische Systeme eine Zustandsgröße existiert, die in allen miteinander im Gleichgewicht befindlichen Systemen übereinstimmt. Die Größe ist die Temperatur T.
1. Hauptsatz der T.: Er stellt eine Ausdehnung des Energieerhaltungssatzes der klassischen Mechanik auf thermodynamische Systeme unter Einbeziehung der Wärme als Energieform dar. Voraussetzung für die Aufstellung des 1. Hauptsatzes war die Erkenntnis, daß Wärme eine Energieform ist und daß Wärme und mechanische Energie wechselseitig in einem festen Verhältnis, dem mechanischen Wärmeäquivalent, ineinander umgewandelt werden können (J. R. Mayer 1842). Analoges gilt für die elektrische Energie (J. P. Joule 1841). H. v. Helmholtz formulierte 1847 das Gesetz von der Erhaltung der Energie, das als allgemeine Form des 1. Hauptsatzes der T. aufgefaßt werden kann: Energie kann weder aus dem Nichts erzeugt, noch in Nichts zerstört werden. Es ist lediglich die Umwandlung einer Form der Energie in eine andere möglich.
Daraus folgt die Unmöglichkeit eines Perpetuum mobile 1. Art, d. h. einer Maschine, die dauernd mehr Energie (z. B. mechanische Arbeit) abgibt als ihr (z. B. in Form von Wärme) zugeführt wird.
Eine andere Formulierung des 1. Hauptsatzes lautet: In einem abgeschlossenen System ist die Summe aller Energien konstant.
Die Gesamtheit des Energievorrates im Inneren des Systems wird als innere EnergieU bezeichnet. Änderungen der inneren Energie sind gleichbedeutend mit Zustandsänderungen des Systems. Sie können erfolgen durch den Austausch von Wärme Δq und/oder Arbeit Δw mit der Umgebung gemäß ΔU = Δw + Δq oder bei sehr kleinen, differentiellen Änderungen dU = ∂w + ∂q. Dabei ist die Vorzeichenkonvention in der chem. T. zu beachten: Dem System zugeführte Größen erhalten positive, vom System abgegebene Größen negative Vorzeichen. Arbeit und Wärme hängen von dem Weg ab, auf dem eine Zustandsänderung erfolgt und sind keine Zustandsfunktionen. In der differentiellen Schreibweise werden ihre Änderungen deshalb mit ∂w und ∂q im Gegensatz zu dU bezeichnet.
Anwendungen des 1. Hauptsatzes.
a) Reine homogene Stoffe: In einem geschlossenen System (Masse m oder Stoffmenge n ist konstant) ist der Zustand eindeutig durch die Zustandsvariablen p, V und T festgelegt. Diese drei Größen sind durch die thermische Zustandsgleichung verknüpft, so daß es genügt, die innere Energie als Funktion von zwei dieser Variablen darzustellen.
Man wählt U = U(T, V). Eine weitere Zustandsgröße ist die EnthalpieH = U + pV. Für sie wird aus Gründen der einfachen Darstellung die Abhängigkeit H = H(T, p) gewählt.
Änderungen der Zustandsgrößen sind wegunabhängig, also totale Differentiale: dU = (∂U/∂T)V dT + (∂U/∂V)T dV, dH = (∂H/∂T)P dT + (∂H/∂p)T dp. Da reine Stoffe Arbeit nur in Form von Volumenarbeit dw = -p dV austauschen können, gilt weiterhin dU = -p dV + ∂q und dH = dU + p dV + V dp = V dp + ∂q. Daraus folgt für isochore Prozesse (V = konst.) dU = ∂q, für isobare Prozesse (p = konst.) dH = ∂q.
Wird von einem geschlossenen System mit seiner Umgebung Wärme ausgetauscht, so ist diese für Vorgänge bei konstantem Volumen gleich der Änderung der inneren Energie, für Vorgänge bei konstantem Druck gleich der Änderung der Enthalpie. Für die partiellen Differentialquotienten in den beiden totalen Differentialen lassen sich folgende Beziehungen ableiten: (∂U/∂T)V = cV = nCV, (∂H/∂T)p = cp = nCp, (∂U/∂V)T = T(∂p/∂T)V – p, (∂H/∂p)T = V – T (∂V/∂T)p. Hierbei bedeuten n Stoffmenge, cV Wärmekapazität bei konstantem Volumen und cp Wärmekapazität bei konstantem Druck, CV und Cp die jeweiligen molaren Wärmekapazitäten.
Die Differentialquotienten (∂p/∂T)V und (∂V/∂T)p sind aus der thermischen Zustandsgleichung zugänglich. So folgt z. B. aus der idealen Gasgleichung (dU/dV)T = 0 und (∂H/∂p)T = 0 (2. Gay-Lussacsches Gesetz), d. h., die innere Energie und die Enthalpie idealer Gase sind unabhängig von Volumen bzw. Druck. Für reale Gase gilt (∂U/∂V)T>< 0 und (∂H/∂p)T>< 0 (Joule-Thomson-Effekt).
b) Zweiphasensysteme von reinen Stoffen: Als zusätzliche Variable tritt hier die Stoffmenge in beiden Phasen auf. Es gelten die Funktionen U = U(T, V, n) und H = H(T, p, n) und für H das totale Differential dH = (∂H/∂T)p,ndT + (∂H/∂p)T,ndp + (∂H/∂n)T,pdn. Der Differentialquotient (∂H/∂n)T,p gibt die Änderung der Enthalpie an, wenn bei konstanter Temperatur und konstantem Druck ein Mol des Stoffes aus einer Phase in eine andere überführt wird: (∂H/∂n)T,p = ΔpH (molare Phasenumwandlungsenthalpie, Phasenumwandlungswärme).
c) Chem. Reaktionen: Bei einer chem. Reaktion enthält das thermodynamische System mehrere Stoffe. Da die innere Energie und die Enthalpie die Summen aller Teilenergien eines Systems sind, gilt im Idealfall U = U1 + U2 + ... = Σ Ui, H = H1 + H2 + ... = Σ Hi, wobei i den Index zur Kennzeichnung der verschiedenen Komponenten bedeutet. Eine Mischung, für die diese Additivität gilt, wird ideale Mischung genannt. Bei realen Mischungen treten zwischen den Teilchen der verschiedenen Komponenten zusätzliche Wechselwirkungen auf, dann gelten diese Additivitätsbeziehungen nicht.
Für die allgemeine chem. Reaktion |νA| A + |νB| B + ... ![]()
νQP + νPQ + ... folgt Σ νiUi = ΔRU und Σ νiHi = ΔRH. Hierbei sind ΔRU und ΔRH die molare Reaktionsenergie bzw. die molare Reaktionsenthalpie. Die experimentelle Bestimmung und die Berechnung von Reaktionsenergien und -enthalpien ist Gegenstand der Thermochemie.
2. Hauptsatz der T. (Entropiesatz): Der 2. Hauptsatz der T. macht Aussagen über die Richtung der Naturvorgänge. Er definiert die EntropieS als neue Zustandsgröße zur Kennzeichnung dieser Richtungsabhängigkeit. Es existieren verschiedene Formulierungen des 2. Hauptsatzes:
1) Wärme kann niemals spontan, d. h. ohne Arbeitszufuhr von außen, von einem kälteren auf einen wärmeren Körper übergehen (Clausius).
2) Es ist unmöglich, eine periodisch arbeitende Maschine zu konstruieren, die weiter nichts bewirkt als die Erzeugung mechanischer Arbeit unter Abkühlung eines Wärmereservoirs (Planck und Thomson). Eine solche nicht realisierbare Maschine wird Perpetuum mobile II. Art genannt. Es wäre eine Wärmekraftmaschine, die in einer Folge von Kreisprozessen (periodisch) Wärme von einem bestimmten Temperaturniveau unter Beachtung des 1. Hauptsatzes ständig in nutzbare Arbeit umwandeln würde.
3) Die Entropie S ist eine Zustandsgröße. In einem abgeschlossenen System kann die Entropie niemals kleiner, sondern nur größer werden (bei irreversiblen Prozessen) oder konstant bleiben (bei reversiblen Prozessen).
Mathematisch formuliert lautet die Aussage des zweiten Hauptsatzes der Thermodynamik dS > 0 für freiwillige, d. h. irreversible Prozesse, dS = 0 im Gleichgewicht und bei reversiblen Vorgängen. Dies gilt nur für abgeschlossene Systeme. In der Praxis spielen jedoch Systeme, die im Energieaustausch mit der Umgebung stehen, eine wichtige Rolle. In diesen Fällen muß die Entropieänderung in der Umgebung stets mit in Rechnung gesetzt werden.
Bei chemischen Reaktionen unter isothermen Bedingungen wird die Reaktionsenthalpie ΔRH vollständig an die Umgebung abgeführt und erhöht deren Entropie um ΔSaußen = -ΔRH/T. Die Gesamtänderung ΔSgesamt des abgeschlossenen Systems, bestehend aus reagierendem System und Umgebung, ergibt sich zu ΔSgesamt = ΔSinnen + ΔSaußen, mit ΔSinnen für die Entropieänderung im reagierenden System. Für ΔSgesamt > 0 verläuft der Vorgang freiwillig.
Weiterhin wurden die freie EnergieF = U – TS und die Gibbssche freie Enthalpie G = H – TS definiert. Diese Zustandsgrößen haben den Vorteil, daß sie auf geschlossene Systeme anwendbar sind, ohne daß die durch den Energieaustausch in der Umgebung resultierenden Veränderungen berücksichtigt werden müssen.
Mit den Funktionen S und G bzw. F läßt sich ein geschlossener Formalismus der Aussagen des 2. Hauptsatzes der T. entwickeln.
Anwendung des 2. Hauptsatzes.
a) Reine Stoffe: Die Entropie ist analog der Zustandsgrößen des 1. Hauptsatzes eine Funktion von p, V und T gemäß S = f(T, V) bzw. S = f(T, p). Beispielsweise gilt für ein ideales Gas dS = nCVd ln T + nR d ln V bzw. dS = nCpd ln T + nR d ln p.
Besteht das System aus zwei Phasen, so ist die Zustandsfunktion S zusätzlich von den Phasenanteilen abhängig: S = f(T, p, n), wobei n die Stoffmenge der reinen Komponenten in einer der beiden Phasen angibt. Das totale Differential enthält dann zusätzlich den partiellen Differentialquotienten (∂S/∂n)T,p = ΔPS. Die molare Phasenumwandlungsentropie ΔPS gibt an, um welchen Wert sich die Entropie des Stoffes (statistisch gesprochen der "Ordnungszustand") ändert, wenn 1 mol aus einer Phase in die andere übergeht. Es besteht der Zusammenhang ΔPS = ΔPH/TP, wobei ΔPH die molare Phasenumwandlungsenthalpie und TP die Temperatur der Phasenumwandlung bezeichnen.
b) Mischungen. Mischungsvorgänge sind wie alle spontanen Prozesse in der Natur irreversibel. Auch bei der Herstellung einer idealen Mischung muß deshalb gelten: ΔSM = S2 – S1 > 0, wenn S1 die Summe der Entropien aller Stoffe vor und S2 diejenige nach dem Mischvorgang darstellt. ΔSM wird Mischungsentropie genannt. Es gilt ΔSM = -R Σ ni ln xi. Dabei sind ni die Stoffmenge und xi der Molenbruch der i-ten Komponente und R die Gaskonstante. Da stets xi< 1 ist, folgt ΔSM > 0. Die mittleren molaren Mischungsfunktionen der inneren Energie U und der Enthalpie H sind demgegenüber für ideale Mischungen Null (1. Hauptsatz).
In realen Mischungen treten aufgrund der Wechselwirkungskräfte zusätzliche Änderungen der Entropie auf, die durch die Verwendung der Aktivitäten ai = fixi (fi = Aktivitätskoeffizient) anstelle der Molenbrüche xi berücksichtigt werden. Für die freie Energie und die freie Enthalpie gelten ähnliche Beziehungen.
c) Reversible Arbeit und Gleichgewichtsbedingungen. Ist ein Prozeß mit einer Änderung der Enthalpie um dH verbunden, so kann von dieser Energiedifferenz der maximal mögliche Anteil dann als Arbeit dwrev gewonnen werden, wenn der Prozeß reversibel durchgeführt wird: dH = dwrev + dqrev. Jeder irreversible Teilschritt (z. B. Reibungs- oder Wärmeleitungsanteil) verschiebt die Aufspaltung von dH zuungunsten des Arbeitsanteiles. Die reversible Arbeit dwrev entspricht der Änderung dG der freien Enthalpie G ≡ H – TS = U + pV – TS oder bei isochorer Prozeßführung der Änderung dF der freien Energie F ≡ U – TS.
Vorgänge verlaufen freiwillig, wenn sie bei reversibler Gestaltung Arbeit leisten, d. h. nach außen abgeben können (dG< 0). Richtungsumkehr bedeutet Vorzeichenumkehr. Unter Arbeitszufuhr (dG > 0) wird dann ein Vorgang erzwungen, der spontan nicht erfolgt. Beispiele sind die Wärmeübertragung von einem Reservoir niedriger auf ein solches höherer Temperatur unter Zuführung elektrischer Arbeit im Kühlschrank oder das Laden eines Akkumulators. Ist die reversible Arbeitsfähigkeit Null, ist das thermodynamische Gleichgewicht erreicht. Zusammengefaßt gelten folgende Kriterien:
dG< 0, dF< 0, dS > 0 freiwillige Vorgänge
dG > 0, dF > 0, dS< 0 unter Arbeitsaufwand
erzwungene Vorgänge
dG = 0, dF = 0, dμi = 0, thermodynamisches
dS = 0 Gleichgewicht.
d) Chem. Reaktionen. Analog wie bei den Zustandsgrößen des 1. Hauptsatzes der T. und entsprechend den dort gegebenen Konventionen ergeben sich für den Ablauf der Reaktion |νA|A + |νB|B + ... ![]()
νpP + νqQ + ...bei konstanter Temperatur und konstantem Druck die Änderungen von G und S je mol Formelumsatz zu ΔG ≡ ΔRG = Σ νiG-i = Σ νiμi und ΔS ≡ ΔRS = Σ νiS-i wobei G-i, und S-i die partiellen molaren freien Enthalpien und Entropien in der Reaktionsmischung sind (partielle molare Größen). ΔRG wird als molare freie Reaktionsenthalpie, ΔRS als molare Reaktionsentropie bezeichnet. Zwischen beiden Größen besteht aufgrund der Definition von G der Zusammenhang ΔRG = ΔRH – TΔRS (Gibbs-Helmholtzsche Gleichung). ΔRG ist der Teil der Reaktionsenthalpie, der bei reversibler isotherm-isobarer Durchführung der chem. Reaktion als Arbeit gewonnen werden kann. Deshalb wird ΔRG auch maximale Nutzarbeit genannt. Eine chem. Reaktion verläuft freiwillig immer in die Richtung, die das System zur Abgabe von Nutzarbeit (ΔRG< 0) befähigt, d. h. -ΔRG ist das thermodynamische Maß für die Affinität chem. Reaktionen.
Führt man die chem. Potentiale μi = μi0 + RT ln ai ein, erhält man ΔRG = Σ νiμi0 + Σ νiRT ln ai = ΔRG0 + RT Σ νi ln ai(van't-Hoffsche Reaktionsisotherme), wobei μi0 das chem. Potential des Stoffes i unter Standardbedingungen (d. h. im reinen Zustand, dem Standardzustand), ai die Aktivität des Stoffes i in der Mischphase und ΔRG0 = Σ νiμi0 die freie Standardeaktionsenthalpie ist.
Im Gleichgewicht gilt ΔRG = 0. Daraus folgt ΔRG0 = -RT Σ νi ln ai oder Π aiνi = e-ΔRG0/RT = K. Die letzte Beziehung ist die thermodynamische Fassung des Massenwirkungsgesetzes. Die Beziehung ΔRG0 = -RT ln K ermöglicht die thermodynamische Berechnung von Gleichgewichtskonstanten K aus freien Standardreaktionsenthalpien. Letztere können thermodynamischen Tabellenwerten direkt entnommen oder über die Gibbs-Helmholtzsche Gleichung ΔRG0 = ΔRH0 – TΔRS0 erhalten werden.
3. Hauptsatz der T. (Nernstsches Wärmetheorem): Aus zahlreichen kalorischen Messungen bei tiefen Temperaturen folgerte Nernst 1906: Bei Annäherung an den absoluten Nullpunkt geht die Änderung ΔS der Entropie eines reinen Stoffes, der sich im inneren Gleichgewicht befindet, gegen Null: ![]()
. Die Entropie nähert sich asymptotisch
einem konstanten Wert S0 der Nullpunktsentropie, die man durch Konvention gleich Null setzen kann (Planck): ![]()
. Daraus folgt weiterhin:
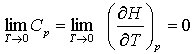
Aus der Sicht der statistischen Deutung der Entropie S = k ln W (k ist die Boltzmann-Konstante, W die thermodynamische Wahrscheinlichkeit) bedeutet S = 0, daß W = 1 ist, d. h., daß das System nur eine Anordnungsmöglichkeit hat, weil sich alle Bestandteile im Grundzustand befinden (vgl. auch Abschnitt über statistische Thermodynamik). Bei sehr tiefen Temperaturen werden jedoch Quanteneffekte wirksam, und eine genaue Behandlung ist nur mit der Quantenstatistik möglich.
Da für T → 0 auch Cp → 0 geht, rufen kleinste Wärmen endliche Temperaturänderungen hervor. Da eine absolute Wärmedämmung aber praktisch unmöglich ist, kann ein System niemals auf T = 0 K abgekühlt werden (Satz von der Unerreichbarkeit des absoluten Nullpunkts). Außerdem ermöglicht der 3. Hauptsatz die Berechnung molarer Standardentropien reiner Stoffe 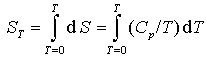
, da S0 = 0. Erfolgen im Temperaturintervall 0 bis T Phasenumwandlungen, so müssen die Phasenumwandlungsentropien dazu
addiert werden. 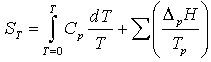
Molare Standardentropien sind in Tabellenwerten zusammengestellt.
II) T. irreversibler Prozesse. Diese ist ebenso wie die phänomenologische T. eine Theorie makroskopischer Systeme, behandelt jedoch Prozesse in Systemen, die sich nicht im Gleichgewicht befinden. Sie beschreibt quantitativ den zeitlichen Ablauf von Transportvorgängen, Ausgleichsprozessen und chem. Reaktionen. Eine mikroskopische Fundierung der T. irreversibler Prozesse ist mit Hilfe der Nichtgleichgewichtsstatistik (kinetische Theorie) möglich. Man unterscheidet die lineare und die nichtlineare T. irreversibler Prozesse.
1) Die lineare T. irreversibler Prozesse gilt in der Nähe des Gleichgewichtes. Aussagen über Gesetzmäßigkeiten der chem. Reaktionskinetik sind mit ihr nur ganz begrenzt möglich, da chem. Reaktionen in der Regel fern vom Gleichgewicht beginnen und die Zeitgesetze stark nichtlinear sind.
Diese T. charakterisiert einen Prozeß durch einen Fluß J, z. B. einen Teilchenstrom bei der Diffusion oder einen Wärmestrom bei der Wärmeleitung. Ursache des Flusses sind Kräfte X, d. s. Abweichungen bestimmter Potentiale oder ihnen proportionaler Größen vom thermodynamischen Gleichgewicht. Die irreversible T. beruht auf drei wesentlichen Postulaten:
a) Die Flüsse Ji sind linear von allen Kräften Xk abhängig, die sie verursachen: ![]()
. Die Proportionalitätskoeffizienten Lik sind die Transportkoeffizienten. Beispiele sind das 1. Ficksche Gesetz der Diffusion und das Gesetz der Wärmeleitung.
b) In Nichtgleichgewichtszuständen tritt an die Stelle der Entropie ihre zeitliche Änderung, die Entropieproduktion P = dS/dt > 0. Sie ist für jeden irreversiblen Prozeß positiv. Das System entwickelt sich so lange, bis es den Gleichgewichtszustand erreicht hat. Dann hat die Entropie ein Maximum erreicht, ihre Änderung und damit auch die Entropieproduktion erreichen den Wert Null. Für den Zusammenhang zwischen Entropieproduktion, Kräften und Flüssen gilt ![]()
c) Bei der Überlagerung mehrerer Vorgänge gilt für die Transportkoeffizienten die Onsagersche Reziprozitätsbeziehung Lik = Lki. Beispielsweise sind die Koeffizienten der Thermodiffusion und des Diffusionsthermoeffektes einander gleich.
2) Die nichtlineare T. irreversibler Prozesse befindet sich noch im Stadium der Entwicklung. Da in großer Entfernung vom Gleichgewicht der lineare Zusammenhang zwischen Kräften und Flüssen nicht mehr gilt, besteht die Möglichkeit zur Strukturbildung in offenen irreversiblen Systemen (dissipative Strukturen). Fragen der Strukturbildung, der Stabilität, periodischer Vorgänge und der Evolution in solchen Systemen werden insbesondere wegen ihrer Bedeutung für die Biochemie und die Reaktionskinetik intensiv bearbeitet.
III) Statistische T. Ihr Ziel ist die Berechnung makroskopischer thermodynamischer Stoffwerte und Zustandsgrößen aus Moleküldaten, den molekularen Bewegungen und Wechselwirkungen. Dabei beschränkt sie sich auf Systeme, die sich im thermodynamischen Gleichgewicht befinden (Gleichgewichtsstatistik).
Aufgrund der Vielzahl der Teilchen in einem makroskopischen System lassen sich die Größen nur bestimmen, wenn Methoden der Wahrscheinlichkeitsrechnung und der mathematischen Statistik in Verbindung mit wenigen Annahmen über die molekularen Eigenschaften herangezogen werden. Die Eigenschaften der Teilchen, z. B. Ort, Geschwindigkeit oder Energie, werden nicht durch diskrete Werte für jedes Teilchen, sondern durch Verteilungsfunktionen beschrieben. Eine Verteilungsfunktion gibt die Wahrscheinlichkeit an, mit der die entsprechende Eigenschaft im System anzutreffen ist. Die makroskopische physikalisch meßbare Größe ergibt sich dann durch statistische Mittelwertbildung, d. h. durch Summation oder Integration über die Funktion.
Für die Frage nach dem thermodynamischen Gleichgewicht spielt die thermodynamische Wahrscheinlichkeit W eine wichtige Rolle. Darunter versteht man die Anzahl der Mikrozustände, durch die ein Makrozustand realisiert werden kann. Als Makrozustand bezeichnet man den Zustand eines makroskopischen Systems, der durch Angaben von Druck, Temperatur und innerer Energie charakterisiert ist. Jede der Anordnungsmöglichkeiten der einzelnen Moleküle des Systems nennt man einen Mikrozustand. W ist im Gegensatz zum üblichen Wahrscheinlichkeitsbegriff eine ganze Zahl und im allg. wesentlich größer als 1. Das System strebt den Makrozustand mit der größten thermodynamischen Wahrscheinlichkeit an. Er entspricht dem Gleichgewichtszustand. Zur Entropie besteht der Zusammenhang S = k ln W (k ist die Boltzmann-Konstante).
Ein Problem besteht in der Abzählung der möglichen Mikrozustände, da durch sie die Art der Verteilungsfunktion bestimmt wird. In der klassischen Mechanik sind die einzelnen Teilchen unterscheidbar und in beliebiger Anzahl in einem Energiezustand unterzubringen. Austausch von zwei Teilchen ergibt einen neuen Mikrozustand. Diese Art der Abzählung liefert die Boltzmann-Verteilung. In der Quantenmechanik sind die einzelnen Teilchen aufgrund der Unschärferelation nicht unterscheidbar. Eine Vertauschung führt nicht zu einem neuen Mikrozustand. Außerdem gilt für Teilchen mit halbzahligem Spin (Fermionen), z. B. Elektronen, das Pauli-Prinzip, d. h., in einem Energiezustand kann nur ein Teilchen untergebracht werden. Das führt zur Fermi-Dirac-Statistik. Teilchen mit ganzzahligem Spin (Bosonen) sind wie klassische Partikeln in beliebiger Anzahl in einem Zustand möglich. Diese Abzählung nennt man Bose-Einstein-Statistik.
Aus der Verteilungsfunktion ergibt sich die Zustandssumme. Sie ermöglicht die Berechnung der thermodynamischen Potentiale, der anderen thermodynamischen Parameter und der Zustandsgleichungen.






Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.