Lexikon der Chemie: Woodward-Hoffmann-Regeln
Woodward-Hoffmann-Regeln, von R. B. Woodward und R. Hoffmann (1965) auf der Grundlage der Molekülorbitaltheorie aufgestelltes Konzept, wonach der Verlauf von Synchronreaktionen wesentlich durch die Erhaltung der Orbitalsymmetrieeigenschaften von Reaktanden und Produkten bestimmt ist. Durch Charakterisierung des Symmetrieverhaltens der an der Reaktion beteiligten Orbitale für den Ausgangs- und Endzustand läßt sich ein Korrelationsdiagramm konstruieren, indem die Energieniveaus von Orbitalen gleicher Symmetrie durch Linien verbunden werden. Aus der Lage der bindenden und lockernden Energieniveaus zwischen Ausgangs- und Endzustand können somit näherungsweise Rückschlüsse auf die Energie des Übergangszustandes gezogen und damit Aussagen über den Reaktionsablauf getroffen werden. Im folgenden werden die W. an ausgewählten Reaktionen dargestellt.
;)
Woodward-Hoffmann-Regeln. Abb. 1: Annäherung von zwei parallelen Ethenmolekülen
;)
Woodward-Hoffmann-Regeln. Abb. 2: Korrelationsdiagramm für die Bildung von Cyclobutan aus zwei Ethenmolekülen. SA symmetrisch bezüglich m1, antisymmetrisch bezüglich m2; AS umgekehrt, SS symmetrisch bezüglich m1 und m2; AA antisymmetrisch bezüglich m1 und m2.
Cycloaddition. Bei der Reaktion von zwei Ethenmolekülen zu Cyclobutan (Cycloaddition) existieren für Ausgangs- und Endzustand zwei Spiegelebenen m1 und m2 (Abb. 1). Bei der Reaktion entstehen aus zwei π-Bindungen zwei σ-Bindungen vom sp3-sp3-Typ (Hybridisierung). Da die entsprechenden Molekülorbitale π1, π2, π1*, π2* bzw. σ1, σ2, σ1*, σ2* den Symmetrieanforderungen des Systems nicht vollständig genügen, werden geeignete Linearkombinationen gebildet. Die so gewonnenen symmetriegerechten Orbitale für Ausgangs- und Endzustand sowie ihr Symmetrieverhalten bezüglich m1 und m2 sind im Korrelationsdiagramm mit aufgeführt (Abb. 2). Beim Übergang vom Ausgangs- in den Endzustand ist eine hohe Aktivierungsenergie erforderlich, da eine Kreuzung der Energieniveaus von bindenden und lockernden Orbitalen auftritt. Somit ist eine Synchronreaktion von zwei im Grundzustand befindlichen Ethenmolekülen zu Cyclobutan ([2 + 2]-Cycloaddition) unter Erhaltung der Orbitalsymmetrie nicht möglich. Durch photochemische Anregung eines Elektrons aus einem π- in ein π*-Orbital ist die Energiebilanz für die Reaktion günstiger, und Ethen dimerisiert zu Cyclobutan. Sieht man die [2 + 2]-Cycloaddition als thermisch verboten und photochemisch erlaubt an, so führen analoge Betrachtungen für die Reaktion von Butadien und Ethen (Diels-Alder-Reaktion) zu der Aussage, daß die [4 + 2]-Cycloaddition thermisch erlaubt und photochemisch verboten ist. Aussagen über die Aktivierungsenergie und damit über den Ablauf von Cycloadditionen können auch im Rahmen der FO-Methode (Abk. von engl. frontier orbital, ›Grenzorbitale‹) von Fukui (1966) erhalten werden. Unter frontier orbitals versteht man das höchste besetzte Molekülorbital (HOMO, Abk. von engl. highest occupied molecular orbital) und das niedrigste unbesetzte Molekülorbital (LUMO, Abk. von engl. lowest unoccupied molecular orbital) eines Moleküls. In Abb. 3 sind die Grenzorbitale des Ethens (H1, L1) und des Butadiens (H2, L2) dargestellt. Bindende Wechselwirkungen im Sinne einer
;)
Woodward-Hoffmann-Regeln. Abb. 3: [4 + 2]-Cycloaddition.
[4 + 2]-Cycloaddition sind aufgrund des Vorzeichens der Molekülorbitale nur zwischen H1 – L2 und H2 – L1 möglich. Als Folge resultiert eine energetische Absenkung der bindenden Orbitale H1 und H2 sowie eine Erhöhung der lockernden Orbitale L1 und L2. Es tritt eine Kreuzung der Energieniveaus der Grenzorbitale auf, was auf eine niedrige Aktivierungsenergie für die thermische [4 + 2]-Cycloaddition schließen läßt. Bisher wurde angenommen, daß der Angriff der Reaktionspartner suprafacial (s) erfolgt, d. h., die gebildeten Bindungen liegen auf der gleichen Seite des reagierenden Systems; beim antarafacialen (a) Angriff liegen die gebildeten Bindungen auf entgegengesetzten Seiten (Abb. 4). Berücksichtigt man diese verschiedenen Angriffsmöglichkeiten, so lassen sich für den Ablauf der Cycloaddition zwischen einem System mit m π-Elektronen und einem mit n π-Elektronen die in der Tab. aufgeführten Regeln angeben. Die Aussagen gelten im gleichen Maße für die Rückreaktion (Cycloreversion).
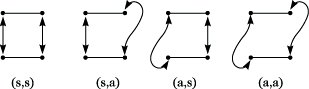
Woodward-Hoffmann-Regeln. Abb. 4: Angriffsmöglichkeiten bei Cycloadditionen. s suprafacial, a antarafacial.
;)
Woodward-Hoffmann-Regeln. Abb. 5: Elektrocyclische Reaktionen.
Elektrocyclische Reaktionen. Sie sind dadurch definiert, daß sich zwischen den Enden eines Polyensystems eine Einfachbindung bildet. Für den Ringschluß müssen die endständigen pπ-Orbitale so gedreht werden, daß eine σ-Bindung ausgebildet werden kann. Die Reaktion kann konrotatorisch oder disrotatorisch ablaufen (Abb. 5). Beide Reaktionswege sind aufgrund ihrer Stereospezifität anhand von Markierungsexperimenten unterscheidbar. Aus der Art der Wechselwirkung von HOMO bzw. LUMO des π-Systems bei der jeweiligen Drehung und der damit verbundenen Veränderung der Grenzorbitalniveaus kann auf den Reaktionsablauf geschlossen werden (Abb. 6). Die konrotatorische Drehung liefert aufgrund des Vorzeichens der Orbitallappen an den endständigen Atomen für das HOMO eine bindende und für das LUMO eine lockernde
;)
Woodward-Hoffmann-Regeln. Abb. 6: Grenzorbitale bei der konrotatorischen Cyclisierung von Butadien.
Wechselwirkung. Damit kann erklärt werden, daß die Cyclisierung von Butadien thermisch auf konrotatorischem Wege erfolgt. Die Verallgemeinerung dieses Konzeptes führt zu der Regel: Thermische elektrocyclische Reaktionen von Systemen mit k π-Elektronen verlaufen disrotatorisch, wenn k =
4 q + 2, und konrotatorisch, wenn k = 4q (q = 0, 1, 2 ...). Bei photochemischen elektrocyclischen Reaktionen kehren sich die Verhältnisse um.
Sigmatrope Reaktionen. Eine sigmatrope Reaktion ist die intramolekulare Wanderung einer von einem oder mehreren π-Elektronensystemen flankierten
σ-Bindung. Die sigmatrope Wanderung, z. B. eines Wasserstoffatoms, kann auf zwei topologisch unterschiedlichen Wegen ablaufen. Bei dem suprafacialen Prozeß befindet sich das wandernde Wasserstoffatom immer auf derselben Seite des π-Systems, während es im antarafacialen Fall von der Oberseite zur Unterseite des π-Systems gelangt. Unter einer cheletropen Reaktion versteht man einen Prozeß, bei dem durch Einfügen eines Atoms zwischen die Enden eines offenkettigen geradzahligen Polyens ein ungeradzahliger Ring gebildet wird oder umgekehrt ein cyclisches Addukt in ein Polyen überführt wird. Ein Beispiel dafür ist der photolytische Zerfall von 3,5-Cycloheptadienon in Hexatrien und Kohlenmonoxid (Abb. 7). Der Ablauf von sigmatropen und cheletropen Reaktionen kann ebenfalls mit Hilfe der W. erklärt werden.
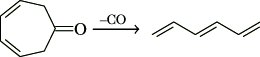
Woodward-Hoffmann-Regeln. Abb. 7: CO-Abspaltung aus 3,5-Cycloheptadienon.
Neben den Korrelationsdiagrammen und der Grenzorbitalbetrachtung gibt es einen weiteren Ansatz zur Abschätzung der Energie des Übergangszustandes in den obigen Reaktionen, der auf Evans (1939) zurückgeht. Das Evans-Prinzip beruht auf der Betrachtung von Aromatizitätseigenschaften cyclischer Übergangszustände in pericyclischen Reaktionen. Bei diesen Reaktionen kann der Übergang von der Ausgangs- in die Endstruktur formal durch Verschiebung von Bindungselektronenpaaren dargestellt werden, wie bei der Cope-Umlagerung oder der Diels-Alder-Reaktion. Thermische pericyclische Reaktionen verlaufen bevorzugt über aromatische Übergangszustände. Photochemische pericyclische Reaktionen führen hingegen bevorzugt zu Produkten, die thermisch über antiaromatische Übergangszustände gebildet werden müßten. Aufgrund der hohen Stereospezifität der Synchronreaktionen haben die W. besonders in der Naturstoffchemie große Bedeutung erlangt.
Woodward-Hoffmann-Regeln. Tab.: Regeln für [m+n]-Cycloadditionen.
| |||
| 4q | [ms + na] [ma + ns] | [ms + ns] [ma + na] | |
| 4q + 2 | [ms + ns] [ma + na] | [ms + na] [ma + ns] |







Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.