Lexikon der Chemie: Alkohole
Alkohole, organische Verbindungen, die Hydroxygruppen -OH an sp3-hybridisierte C-Atome gebunden enthalten. Die A. lassen sich durch formale Substitution von H-Atomen in Kohlenwasserstoffen ableiten, z. B. Alkanole aus Alkanen oder Cycloalkanole aus Cycloalkanen. Das die OH-Gruppe bindende C-Atom kann auch Bestandteil einer Seitenkette von Aromaten, z. B. Benzylalkohol, oder Heteroaromaten, z. B. Furfurylalkohol, sein.
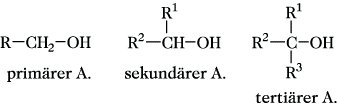
Je nach Anzahl der OH-Gruppen unterscheidet man ein-, zwei-, drei- und mehrwertige A., wobei an jedes C-Atom nur eine OH-Gruppe gebunden ist. Bekannte einwertige A. sind Methanol und Ethanol, einfache zwei- bzw. dreiwertige A. (Diole, Triole) sind Glycole und Glycerin (Tab. 1 und 2). Durch Reduktion von Monosacchariden leiten sich sechswertige A. ab, z. B. D-Glucitol und D-Mannitol. Weiterhin wird unterschieden zwischen primären, sekundären und tertiären A., je nachdem, ob das die OH-Gruppe bindende C-Atom mit einem, zwei oder drei weiteren C-Atomen verknüpft ist. Somit ergeben sich folgende allgemeine Formeln:
Alkohole. Tab. 1: Homologe Reihe einwertiger Alkohole
| ||
| Ethanol | CH3-CH2-OH | |
| Propan-1-ol | CH3-CH2-CH2-OH | |
| Propan-2-ol | CH3-CH(OH)-CH3 | |
| Butan-1-ol | CH3-CH2-CH2-CH2-OH | |
| Butan-2-ol | CH3-CH2-CH(OH)-CH3 | |
| 2-Methylpropan-1-ol (Isobutanol) | CH3-CH-CH2-OH | CH3 | |
| 2-Methylpropan-2-ol (tert-Butanol) | CH3 | CH3-C-OH | CH3 |
Alkohole. Tab. 2: Einige zwei- und mehrwertige Alkohole
| ||
| Propylenglycol (Propan-1,2-diol) | HO-CH2-CH(OH)-CH3 | |
| Propan-1,3-diol | HO-CH2-CH2-CH2-OH | |
| Glycerin (Propan-1,2,3-triol) | HO-CH2-CH(OH)-CH2-OH | |
| Pentaerythrit | CH2-OH | HO-CH2-C-CH2-OH | CH2-OH |
Nomenklatur. Die Bezeichnung der einwertigen A. erfolgt durch Kombination des Namens des Stammkohlenwasserstoffes mit dem Suffix -ol. Zur Unterscheidung von Isomeren wird die Stellung der OH-Gruppe durch eine Ziffer vor der Endsilbe angegeben, z. B. Propan-2-ol. Üblich ist auch noch die Namensbildung durch Anhängen von -alkohol an die Bezeichnung des Radikals, z. B. Methyl-, Ethyl- oder Allylalkohol. Für den einfachsten tertiären A. ergeben sich daher drei Möglichkeiten der Benennung: tert-Butanol, tert-Butylalkohol oder 2-Methylpropan-2-ol. Bei zwei- oder mehrwertigen A. wird analog unter Verwendung der Endungen -diol, -triol usw. verfahren und die Stellung der OH-Gruppen mit möglichst kleinen Ziffern angegeben, z. B. Butan-1,4-diol. Anderenfalls werden die zugelassenen Trivialnamen verwendet.
Eigenschaften. Niedermolekulare A. sind Flüssigkeiten mit typischem Geruch und brennendem Geschmack, die aufgrund von Molekülassoziationen durch intermolekulare Wasserstoffbrückenbindungen relativ hohe Siedepunkte im Vergleich zu Kohlenwasserstoffen oder anderen funktionellen Verbindungen gleicher Molmasse aufweisen. Noch ausgeprägter ist dies bei den viskosen Glycolen oder Glycerin. Bei höheren A. handelt es sich um feste Verbindungen mit nur schwach ausgeprägtem Geruch. Niedere A. sind wegen der Wechselwirkung der hydrophilen polaren OH-Gruppe mit Wassermolekülen in jedem Verhältnis mit Wasser mischbar. Die Grenze der Löslichkeit liegt bei einwertigen A. bei C4, bei mehrwertigen A. wesentlich höher. Von den isomeren Butanolen ist tert-Butanol noch in jedem Verhältnis mit Wasser mischbar, während die isomeren Amylalkohole (C5) praktisch nicht mehr in Wasser löslich sind, da bei ihnen bereits der hydrophobe Charakter der Alkylradikale überwiegt. D-Glucitol oder Mannitol sind in Übereinstimmung mit der Theorie gut in Wasser löslich. Sie zeigen jedoch lipophile Eigenschaften, d. h., sie lösen sich nicht mehr in Diethylether und Ethanol. Viele A. sind charakteristisch physiologisch wirksam (Methanol, Ethanol, Fuselöl).
Niedere einwertige A. sind brennbar und leicht entflammbar (Brennspiritus). A. reagieren gegenüber Säure-Base-Indikatoren neutral, jedoch besteht die Möglichkeit der Salzbildung durch Einwirkung reaktiver Metalle der I. bis III. Hauptgruppe des Periodensystems unter Ausschluß von Wasser. Dabei entstehen durch Substitution des H-Atoms der OH-Gruppe durch Metall Alkoholate.
Die Bildung von Alkoxonium-Ionen, die als Zwischenstoffe bei chemischen Reaktionen auftreten können, erfolgt nur mit starken Brönsted-Säuren oder mit Lewis-Säuren, z. B. BF3:
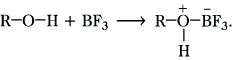
Reaktionen. Typische Reaktionen der A. sind die Veresterungen mit anorganischen Säuren zu Alkylhalogeniden, Alkylnitriten, Alkylnitraten, Alkyl- und Dialkylsulfaten sowie Trialkylphosphaten und mit Carbonsäuren, ihren Anhydriden oder Säurechloriden zu Carbonsäureestern: R-CH2-OH + HBr → R-CH2-Br + H2O, C2H5OH + R-COOH ![]()
R-COOC2H5 + H2O. Die Umsetzung von A. mit 3,5-Dinitrobenzoylchlorid zu gut kristallisierenden 3,5-Dinitrobenzoesäureestern dient zur Identifizierung der A.:
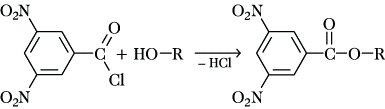
Die frei werdende Salzsäure wird durch Basen neutralisiert.
Beim Erhitzen von Schwefel-, Phosphor- oder Oxalsäure reagieren A. im Verlaufe einer intramolekularen Dehydratisierung zu Alkenen oder einer intermolekularen Wasserabspaltung zu Ethern. Beide Reaktionen konkurrieren miteinander. Hohe Temperaturen begünstigen die Alkenbildung, tiefere die Etherbildung: C2H5OH → CH2=CH2 + H2O, 2 C2H5OH → C2H5-O-C2H5 + H2O.
Analog sind die Verhältnisse bei der Gasphasendehydratisierung an Al2O3. Dabei entstehen bei 350 bis 400 °C fast ohne Nebenprodukte Alkene, bei 260 °C dagegen vorwiegend Ether. In der Reihenfolge primärer A. < sekundärer A. < tertiärer A. steigt die Leichtigkeit der intramolekularen Wassereliminierung (Alkenbildung). Die Etherbildung wird durch Überschuß an A. begünstigt.
Bei der Bildung von Alkenen entsteht das thermodynamisch stabilere Produkt (Saytzeff-Orientie-rung), z. B. bildet sich aus Butan-2-ol But-2-en und nicht But-1-en:
A. können zu Aldehyden, Ketonen und Carbonsäuren oxidiert bzw. dehydriert werden. primäre A. ergeben dabei zunächst Aldehyde, die zu Carbonsäuren weiteroxidiert werden können:
Sekundäre A. werden zu Ketonen oxidiert:
Tertiäre A. werden nicht oxidiert. Die Identifizierung der unterschiedlichen Oxidationsprodukte wird zur analytischen Unterscheidung der primären, sekundären und tertiären A. genutzt. Als Oxidationsmittel verwendet man Kaliumdichromat in Schwefelsäure, Chrom(VI)-oxid, tert-Butylchromat oder Pyridin-Chromsäure-Komplexe. Zur durchgehenden Oxidation primärer A. zu Carbonsäuren eignen sich auch Kaliumpermanganat oder Salpetersäure, zur Umsetzung primärer A. zum Aldehyd aktivierter Braunstein und Selen(IV)-oxid. Primäre und sekundäre A. können auch mit Aceton in Anwesenheit von Aluminiumisopropylat zu Aldehyden bzw. Ketonen umgesetzt werden (Oppenauer-Oxidation). Industriell erfolgt die Dehydrierung zu Aldehyden oder Ketonen mit Luftsauerstoff an Metallkatalysatoren bei 400 bis 600 °C.
Analytisches. Vorproben sind der Nachweis der neutralen Reaktion und gegebenenfalls die Reaktion mit kleinen Portionen metallischen Natriums unter Wasserstoffentwicklung, ferner die Bildung des mit grüner Flamme brennenden Borsäuremethylesters für Methanol und die Iodoformprobe für Ethanol oder Verbindungen mit der Gruppierung CH3-CHOH-. Die Unterscheidung primärer, sekundärer und tertiärer A. ist durch ihre unterschiedlichen Oxidationsprodukte möglich. Die Identifizierung erfolgt als 4-Nitrobenzoesäure- oder 3,5-Dinitrobenzoesäureester über die Umsetzung mit den entsprechenden Säurechloriden bzw. durch Umsetzung mit Arylisocyanat zu den ebenfalls kristallinen Urethanen. In den IR-Spektren finden sich Banden im Bereich von 1050 bis 1300 cm-1 (C-O-Valenzschwingung) sowie von 3200 bis 3700 cm-1 (O-H-Valenzschwingung). Die Lage der O-H-Valenzschwingung ist dabei stark vom Assoziationsgrad und damit von der Konzentration abhängig. Während z. B. freie Hydroxygruppen Absorptionsbanden zwischen 3590 und 3650 cm-1 zeigen, sind sie bei Vorliegen von intramolekularen (3420 bis 3590 cm-1) und intermolekularen Wasserstoffbrückenbindungen (3200 bis 3550 cm-1) nach niederen Wellenzahlen verschoben. Im Kernresonanzspektrum sind die chemischen Verschiebungen für die OH-Protonen in starkem Maße von der Polarität der O-H-Bindung sowie vom Assoziationsgrad abhängig, d. h., diese Spektren sind nicht sehr aussagekräftig. In den Massenspektren zeigen die A. oft nur schwache Molpeaks. Das Fragmentierungsverhalten wird durch Eliminierung von Wasser und Spaltung der C-COH-Bindung bestimmt. Als charakteristisches Bruchstück erscheint das Ion [CH2=OH]+.
Vorkommen. Einzelne A. kommen frei oder gebunden natürlich vor, z. B. Glycerin als Baustein von Fetten, Ölen und Phosphatiden oder höhermolekulare einwertige A., wie Cetyl-, Ceryl- oder Myricylalkohol, in Wachsen pflanzlicher oder tierischer Herkunft. Biologisch interessante Verbindungen sind weiterhin Geraniol und Menthol als Vertreter der Terpene, Farnesol aus der Stoffklasse der Sesquiterpene und Retinol (Vitamin A1) als Diterpen sowie Hydroxyverbindungen aus der Klasse der Steroide, z. B. Cholesterin.
Herstellung. 1) Hydratisierung von Alkenen in Gegenwart von Säuren:
2) Epoxidierung von Alkenen mit anschließender Hydrolyse der Epoxide (Oxirane):
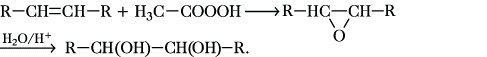
Als Reagenzien werden z. B. Peroxyessigsäure oder Sauerstoff und Silberoxid eingesetzt;
3) Addition von unterchloriger Säure HOCl an Alkene sowie basische Hydrolyse der als Zwischenprodukte entstehenden Chlorhydrine und Epoxide:
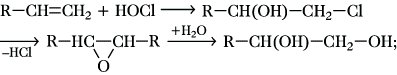
4) Umsetzung von Ethin mit Formaldehyd (Acetylenchemie) mit anschließender katalytischer Hydrierung des erhaltenen Butin-1,4-diols:
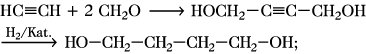
5) Reaktion von Alkylhalogeniden mit Alkalihydroxiden: R-CH2-Cl + OH- → R-CH2-OH + Cl-;
6) Hydrolyse von Carbonsäureestern, die entweder natürlich vorkommen oder aus Alkylhalogeniden darstellbar sind;
7) Reduktion von Carbonsäureestern: a) Bouveault-Blanc-Reduktion. b) Katalytische Hydrierung unter Verwendung von Kupfer(II)-Chrom(III)-oxid-Mischkatalysatoren;:
R1-COOR2 + 4 H → R1-CH2OH + R2-OH;
8) Reduktion von Aldehyden und Ketonen: a) Katalytische Hydrierung mit Raney-Nickel: R-CHO + H2 → R-CH2-OH. b) Reduktion mit Natrium und Ethanol: R-CHO + 2 H → R-CH2-OH.
c) Reduktion mit komplexen Metallhydriden, z. B. Lithiumaluminiumhydrid oder Natriumborhydrid: R-CHO + 2 H → R-CH2-OH. d) Reduktion von Ketonen zu Pinacolen. e) Meerwein-Ponndorf-Verley-Reduktion. f) Cannizzaro-Reaktion;
9) Grignard-Reaktionen mit Aldehyden, Ketonen und Carbonsäureestern: a) Umsetzungen von Formaldehyd mit Grignard-Reagens zu primären A.:
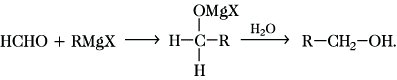
b) Umsetzungen anderer Aldehyde ;ud Ketone sowie Carbonsäureester zu sekundären und tertiären A. (Grignard-Reaktionen);
10) Vollständige katalytische Hydrierung von Phenolen zu Cyclohexanolen unter Druck bei höheren Temperaturen im Autoklaven;
11) Umsetzungen primärer aliphatischer Amine mit salpetriger Säure: R-CH2-NH2 + HNO2 → [R-CH2-N=N-OH] → R-CH2-OH + N2;
12) Oxidation von technisch zugänglichen Aluminumtrialkylen mit nachfolgender Hydrolyse:
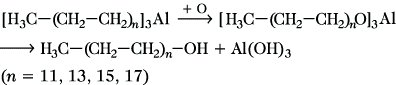
Diese Reaktion ist technisch besonders wichtig für die Gewinnung von langkettigen Alkan-1-olen als Ausgangsstoffe für Tenside.
Verwendung. Niedermolekulare einwertige A. werden in großem Maßstab als Lösungsmittel, als Brennstoffe sowie zur Darstellung von Estern, Ethern und Alkenen eingesetzt. Einfache zwei- und dreiwertige A. werden für Kosmetika, als Gefrierschutzmittel, zur Herstellung von Sprengstoffen, Schmier- und Textilhilfsmitteln u. a. verwendet.





Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.