Lexikon der Chemie: Infrarotspektroskopie
Infrarotspektroskopie, IR-Spektroskopie, Ultrarotspektroskopie, UR-Spektroskopie, Teilgebiet der Spektroskopie, das die Wechselwirkung elektromagnetischer Strahlung aus dem infraroten Spektralbereich mit einer Probe untersucht. Wenn eine geeignete Probe eines Stoffes mit IR-Strahlung durchstrahlt wird, so wird eine Reihe von Frequenzen dieser IR-Strahlung absorbiert, die andere wird durchgelassen. Trägt man die prozentuale Absorption A oder Durchlässigkeit der Probe in Abhängigkeit von der Wellenzahl ν~ oder Wellenlänge auf, so erhält man ihr IR-Spektrum (Abb. 1). Bei der Absorption der Strahlung ändern die Moleküle ihre Rotations- und Schwingungsenergie. Die dabei entsprechend der Gleichung ΔE= h ν (E Energie, h Plancksche Konstante) auftretenden Rotations- und Schwingungsfrequenzen ν erlauben weitgehende Rückschlüsse auf die Struktur von Molekülen und gestatten deren qualitative und quantitative Bestimmung.
;)
Infrarotspektroskopie. Abb. 1: IR-Spektrum von Polystyrol.
Theoretische Grundlagen. Die Atome in einem Molekül sind nicht starr miteinander verbunden, sondern können Schwingungen um ihren Gleichgewichtsabstand ausführen. Diese lassen sich näherungsweise durch die Gesetze der klassischen Physik beschreiben, indem man die Atome durch Kugeln, die Bindungskräfte durch Federn darstellt (Abb. 2). Ein Molekül aus N Atomen hat 3N Freiheitsgrade der Bewegung. Davon fallen 3 Freiheitsgrade auf die Translation, 3 bei nichtlinearen bzw. 2 bei linearen Molekülen auf die Rotation, so daß für nichtlineare Moleküle 3N - 6, für lineare 3N - 5 Schwingungsfreiheitsgrade verbleiben, die gleich der Anzahl der Eigenschwingungen eines Moleküls sind.
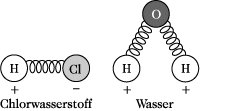
Infrarotspektroskopie. Abb. 2: Mechanische Molekülmodelle.
Die theoretisch mögliche Anzahl von Schwingungen in einem Molekül (3N – 6 bzw. 3N- 5) werden im allg. im IR-Spektrum nicht beobachtet. Es gelten bestimmte Auswahlregeln: Infrarotes Licht wird nur dann absorbiert, wenn ein sich bei einer Molekülschwingung änderndes Dipolmoment mit dem schwingenden elektrischen Vektor der elektromagnetischen Strahlung in Wechselwirkung tritt. Das ist dann der Fall, wenn das Dipolmoment des Moleküls in der einen Extremlage der Schwingung von dem in der anderen Extremlage verschieden ist. Es liegt dann eine IR-aktive Schwingung vor, die im IR-Spektrum beobachtet wird, Schwingungen, für die diese Bedingungen nicht gelten, sind IR-inaktiv. Sie können nur im Raman-Spektrum (Raman-Spektroskopie) beobachtet werden. Eine weitere Vereinfachung der IR-Spektren kann dadurch auftreten, daß bestimmte Schwingungen entartet sind, d. h., daß sie die gleiche Frequenz haben, aber in verschiedenen Raumrichtungen erfolgen, z. B. beim CO2-Molekül (Tab.1). In erster Näherung kann ein solches System – als Beispiel sei ein zweiatomiges Molekül betrachtet – als harmonischer Oszillator behandelt werden, dessen Schwingungsfrequenz sich aus dem Hookeschen Gesetz zu
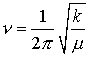
ergibt, wobei ν die Schwingungsfrequenz in Hz, k die Kraftkonstante der Bindung in Nm-1 und
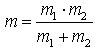
die reduzierte Masse ist.
Infrarotspektroskopie. Tab. 1: Schwingungen des CO2-Moleküls.
;)
Löst man diese Gleichung durch Einsetzen entsprechender Werte, so findet man, daß die entsprechenden Frequenzen in den infraroten Spektralbereich fallen.
Die potentielle Energie eines harmonischen Oszillators (auch Oszillatorpotential genannt) in Abhängigkeit vom Abstand der schwingenden Atome wird durch eine Potentialkurve, die eine Parabel darstellt, beschrieben (Abb. 4). Für einen klassischen harmonischen Oszillator sollte dabei jede Schwingungsamplitude erlaubt sein.
;)
Infrarotspektroskopie. Abb. 3: Potentialurve des klassischen harmonischen Oszillators (x1, x2 beliebige Auslenkungen).
Dies steht jedoch im Widerspruch zur experimentellen Beobachtung diskreter Schwingungsfrequenzen im IR-Spektrum. Eine Lösung dieses Problems war mit Hilfe der Quantenmechanik möglich. Die Schrödinger-Gleichung für einen eindimensionalen harmonischen Oszillator führt zu dem Ergebnis, daß die Gesamtenergie E nur die Werte E = hν(v + 1/2) annehmen kann, wobei v = 0, 1, 2, 3, ... die Schwingungsquantenzahl darstellt. Danach kann ein harmonischer Oszillator nach der Quantenmechanik nur bestimmte Energiewerte annehmen, deren Differenz hν beträgt (Abb. 4). Übergänge von einem niederen zu einem höheren Schwingungsenergieniveau können durch Absorption einer Frequenz erfolgen, die gleich der Frequenz der mechanischen Oszillation ist. Übergänge zwischen nicht benachbarten Energiezuständen sind nach den gültigen Auswahlregeln verboten, ebenso Wechselwirkungen zwischen verschiedenen Schwingungen.
;)
Infrarotspektroskopie. Abb. 4: Quantenmechanischer harmonischer Oszillator.
Aus der quantenmechanischen Berechnung folgt nun das wichtige Resultat, daß die niedrigste mögliche Energie, die Grundzustandsenergie, des harmonischen Oszillators nicht Null ist, sondern den Wert E0=hν/2 hat. Da sich der harmonische Oszillator am absoluten Nullpunkt der Temperatur (T= 0 K) mit Sicherheit im Grundzustand befindet, wird diese Energie auch als Nullpunktsenergie bezeichnet. Der endliche, von Null verschiedene Wert dieser Nullpunktsenergie ist eine Forderung der Heisenbergschen Unbestimmtheitsrelation, da andernfalls bei E0= 0 Ort und Impuls gleichzeitig scharfe Werte, nämlich Null, hätten. Ein klassisches Teilchen hingegen würde bei T = 0 K in seiner Gleichgewichtslage x= 0 ruhen.
Reale Modelle können nur näherungsweise als harmonische Oszillatoren beschrieben werden. Für sie gilt das Modell des anharmonischen Oszillators, das die in Abb. 5 dargestellte Potentialkurve aufweist. Darin haben die verschiedenen Schwingungsenergiezustände nicht mehr gleichen Abstand voneinander, Übergänge zwischen nicht benachbarten Zuständen sind erlaubt, Wechselwirkungen zwischen verschiedenen Schwingungen möglich. Die Energiedifferenz zwischen zwei benachbarten Schwingungsniveaus ist ΔE= hν(1 - 2vx), wobei x die Anharmonizitätskonstante ist, die gewöhnlich einen Wert < 0,05 aufweist. Übergänge zwischen dem Schwingungsgrundzustand (v = 0) und dem zweiten oder höheren Anregungszuständen geben Anlaß zu Oberschwingungen, die bei angenähert der doppelten, dreifachen ... Frequenz der Grundschwingung erscheinen. Oberschwingungen haben eine wesentlich geringere Wahrscheinlichkeit als Grundschwingungen, so daß ihre entsprechende Absorption nur eine geringe Intensität aufweist.
;)
Infrarotspektroskopie. Abb. 5: Potentialkurve des quantenmechanischen anharmonischen Oszillators (schwarz). Zum Vergleich Potentialkurve des harmonischen Oszillators (grau).
Wechselwirkungen zwischen verschiedenen Molekülschwingungen führen zu Kombinationsbanden. Sie resultieren aus einer Absorption von infraroter Strahlung, deren Frequenz gleich der Summe oder Differenz zweier Grundschwingungen ist. Die Wahrscheinlichkeiten der Anregung von Kombinationsschwingungen sind denen der Oberschwingungen vergleichbar. Die bisherigen Ausführungen gelten für reine Schwingungen. Tatsächlich gibt es jedoch kein reines Schwingungsspektrum, sondern jeder Schwingungsübergang ist von Rotationsübergängen begleitet. Da die Energie, die zur Anregung eines Schwingungsüberganges notwendig ist, stets auch zur Anregung von Rotationsübergängen ausreicht, so treten beide zusammen auf, und man spricht korrekter von Rotationsschwingungsspektren (Abb. 6).
;)
Infrarotspektroskopie. Abb. 6: Termschema im Rotationsschwingungsspektrum.
Die beiden ausgezogenen Linien stellen in einem Energiediagramm die beiden niedrigsten Schwingungsniveaus seines Moleküls dar. Zu jedem Schwingungszustand gehört eine Reihe von Rotationsniveaus, die als gestrichelte Linien mit den dazugehörigen Rotationsquantenzahlen J eingezeichnet sind. Bei einer Änderung der Schwingungsquantenzahl von v= 0 nach v = 1 erfolgt unter Berücksichtigung der Auswahlregel ΔJ = ±1 ein Übergang von einem Rotationsniveau des Schwingungsgrundzustandes zum nächsthöheren oder nächstniederen Rotationsniveau des ersten angeregten Schwingungszustandes, wie es durch einige Pfeile dargestellt ist. Man erhält so anstelle einer einzigen "Schwingungslinie" ein ganzes Liniensystem, das in zwei Teile zerfällt. Ein Teil, für den ΔJ = +1 ist, nennt man den R-Zweig, den anderen Teil, für den ΔJ = -1 ist, den P-Zweig. Im R-Zweig addiert sich die Energie des Rotationsüberganges zu der der Schwingung, im P-Zweig wird diese Rotationsenergie von der Schwingungsenergie subtrahiert. Zwischen beiden Zweigen fehlt die 0-0-Linie, die infolge der Auswahlregeln für die Änderung der Rotationsquantenzahl verboten ist. Die Intensitätsverteilung innerhalb beider Zweige entspricht wie bei den reinen Rotationsspektren der thermischen Verteilung der Moleküle auf die Rotationszustände. Die Lage der entsprechenden Absorption im Spektrum wird im wesentlichen durch die Energiedifferenz der Schwingungszustände, die Feinstruktur durch die Änderung der Rotationszustände bestimmt. Eine derartige Anhäufung von Absorptionslinien, die zu einem Schwingungsübergang, aber zu verschiedenen Rotationsübergängen gehören, nennt man eine Absorptionsbande (Abb. 7).
Infrarotspektroskopie. Tab. 2: Bauelemente von IR-Spektrometern.
| |||||||
| Wellenzahl ~ν in cm-1 | 12500 | 4000 | 200 | 10 | |||
| Wellenlänge λ in mm | 0,8 | 2,5 | 50 | 1000 | |||
| Lichtquelle | Wolframband- lampe | Nernst-Stift, SiC-Stift, Keramikstab | Hg-Hoch- druckbrenner | ||||
| optische Material | Quarz | NaCl, KBr, LiF, CsBr | |||||
| dispergierendes System | Prisma Gitter | Prisma Gitter | Gitter | ||||
| Empfänger | PbS- Photozelle | Thermoelement Golayzelle Bolometer | Golayzelle |
;)
Infrarotspektroskopie. Abb. 7: Absorptionsbande im Rotationsschwingungsspektrum des Kohlenmonoxids.
Apparatives. Moderne lichtelektrische Spektralphotometer für den infraroten Spektralbereich arbeiten unter Verwendung geeigneter Bauelemente nach dem Prinzip eines Zweistrahlwechsellichtverfahrens (Spektralapparaturen). Mit derartigen Geräten ist die Aufnahme eines IR-Spektrums in einigen Minuten möglich. Mit Spezialgeräten können Aufnahmezeiten von Bruchteilen von Sekunden erreicht werden. Da das gesamte IR-Gebiet nicht mit einem einzigen Spektrometer vermessen werden kann, wurde das infrarote Spektralgebiet in nahes IR (NIR), mittleres IR und fernes IR (FIR) unterteilt, wobei sich Geräte für diese Gebiete in ihrer apparativen Ausrüstung unterscheiden (Tab. 2).
In neuerer Zeit werden in zunehmendem Maße Fourier-Spektrometer im IR verwendet. Sie weisen eine wesentlich erhöhte Empfindlichkeit gegenüber den üblichen Dispersionsspektrometern auf und erlauben eine schnellere Registrierung der Spektren. Ihr Einsatz empfiehlt sich vor allem in intensitätslimitierten Situationen, z. B. im FIR.
Proben können nach entsprechender Präparation in allen drei Aggregatzuständen vermessen werden. Der Substanzverbrauch zur Aufnahme eines IR-Spektrums ist gering und liegt in der Größenordnung von einigen Milligramm, läßt sich aber mit Spezialausrüstungen noch beträchtlich senken.
Anwendung der IR-Spektroskopie. 1) Das am weitesten verbreitete Anwendungsgebiet ist die Strukturaufklärung (Abb. 8). Von den möglichen Schwingungen in einem Molekül sind einige in erster Näherung in einzelnen Bindungen oder Atomgruppen lokalisiert (lokalisierte Schwingungen), während andere das Molekül als Ganzes einschließen (Gerüstschwingungen).
Die lokalisierten Schwingungen bestehen aus Valenzschwingungen oder aus Deformationsschwingungen. Viele dieser lokalisierten Schwingungen, insbesondere der Valenzschwingungen, können zum Nachweis funktioneller Gruppen dienen. Unterschiedliche Atome besitzen unterschiedliche Massen, unterschiedliche Bindungen verschiedene Kraftkonstanten. Je nach den an einer Schwingung beteiligten Atomen und ihren Bindungszuständen resultieren nach der bereits erwähnten Gleichung
verschiedene Schwingungsfrequenzen. So ist z. B. die C≡C-Bindung schwerer zu dehnen oder zu stauchen als die C=C-Bindung. Diese wiederum ist stärker als die C-C-Bindung. Die Kraftkonstanten von Dreifach-, Doppel- und Einfachbindungen verhalten sich etwa wie 3 : 2 : 1. Ihre Schwingungsfrequenzen stehen dann im Verhältnis
Neben der Kraftkonstanten wird die Schwingungsfrequenz auch durch die reduzierte Masse der beteiligten Atome bestimmt.
Bei k = konst. ist ![]()
;)
Infrarotspektroskopie. Abb. 8: IR-Spektrum von Phthalsäuredimethylester.
Infrarotspektroskopie. Tab. 3: Erwartungsbereiche charakteristischer Frequenzen.
| |||||||
| X-H Valenzschwingungen | X≡Y Valenz- schwin- gungen | X=Y Valenz- schwin- gungen | Deformationsschwingungen, Gerüstschwingungen, Valenzschwingungen schwerer Atome |
Wenn die Masse der beteiligten Atome ansteigt, fällt ihre Schwingungsfrequenz. Wegen der geringen Masse des Wasserstoffs haben alle Schwingungen, an denen H beteiligt ist, extrem hohe Schwingungsfrequenzen. Aus diesen Überlegungen lassen sich bestimmte Wellenzahlbereiche abschätzen, in denen funktionelle Gruppen absorbieren, die oberhalb 1500 cm-1 zu finden sind (Tab. 3). Solche lokalisierten Schwingungen sind für eine Vielzahl funktioneller Gruppen tabellarisch niedergelegt worden. Dabei ist die Ableitung der Erwartungsbereiche überwiegend rein empirisch erfolgt. Bestimmte Atomgruppierungen (z. B. eine -OH- oder eine /C=O-Gruppe) ergeben stets bei bestimmten Wellenzahlen – das können auch mehrere sein – Absorptionen, deren Lage vom Molekülrest weitgehend unabhängig ist. Ist eine solche "charakteristische Frequenz" für eine Atomgruppierung in einer genügend großen Anzahl von bekannten Verbindungen beobachtet worden, so kann man ohne irgendwelche Rechnungen allein aus ihrem Auftreten auf die Gegenwart dieser Atomgruppe in Substanzen unbekannter Struktur schließen. Mit solchen charakteristischen Frequenzen ist also der Nachweis von Atomgruppen und damit die Erschließung der Struktur unbekannter Substanzen möglich.
2) Identifizierung von Substanzen. Aus Tab. 3 folgt, daß ein IR-Spektrum unterhalb 1500 cm-1 relativ bandenreich und unübersichtlich ist. Hier erscheinen Gerüstschwingungen, zahlreiche Deformationsschwingungen, aber auch noch Valenzschwingungen schwerer Atome, z. B. der Kohlenstoff-Halogen-Bindung. Schwingungszuordnungen sind deshalb in diesem Gebiet erschwert. Die IR-Absorptionen sind jedoch für das Molekül als Ganzes sehr charakteristisch, so daß dieses Gebiet auch als Fingerabdruck- (fingerprint-) Gebiet bezeichnet wird, das sich hervorragend zur Feststellung der Identität einer Substanz mit einer authentischen Probe eignet. Der Identitätsnachweis zweier reiner Substanzen wird dadurch geführt, daß man die IR-Spektren beider Stoffe Bande für Bande auf ihre Identität überprüft. Im Gegensatz zu vielen anderen Stoffkonstanten, wie Siedepunkt, Schmelzpunkt, Dichte, Brechung, hat das IR-Spektrum den Vorteil, infolge der vielen in ihm enthaltenen Absorptionsbanden eine wesentlich größere Anzahl von Vergleichsmöglichkeiten zu bieten. Substanzen ähnlicher Struktur werden sich zwar auch in ihrem IR-Spektrum bis zu einem gewissen Grad ähneln, identische Spektren ergeben sie jedoch nicht.
3) Die quantitative IR-Analyse ist in den letzten Jahren durch eine Reihe anderer Methoden (z. B. Gaschromatographie, UV-Spektroskopie) zurückgedrängt worden. Dennoch ist sie für bestimmte Probleme weiterhin wichtig. Dabei erweist sich der Bandenreichtum des IR-Spektrums als vorteilhaft, da dadurch die Auswahl geeigneter Analysenbanden, die möglichst wenig durch Absorptionsbanden der anderen vorhandenen Komponenten beeinflußt werden, erleichtert wird. Es sind auch Mehrkomponentenanalysen durchführbar. Quantitative IR-Bestimmungen lassen sich prinzipiell nach zwei Methoden vornehmen: unter Verwendung von Kalibrierkurven und auf der Grundlage des Lambert-Beerschen Gesetzes.
Die Genauigkeit der Methoden beträgt etwa l %. Dies ist unter anderem abhängig vom Spektrometertyp, den verwendeten Geräteparametern und den spektralen Eigenschaften der zu bestimmenden Substanzen. Eine Verbesserung kann durch die digitale Erfassung der Meßdaten verbunden mit Computerberechnungen erreicht werden.
Bei der Anwendung des Lambert-Beerschen Gesetzes muß unbedingt beachtet werden, daß im IR die Bandenintensität von der spektralen Spaltbreite des Spektrometers abhängig ist. Wahre Werte erhält man nur dann, wenn die spektrale Spaltbreite kleiner als 1/5 der Halbwertsbreite der vermessenen Bande ist. Dies ist im allg. bei Gittergeräten der Fall, bei Prismengeräten nicht.
4) Die Untersuchung von Wasserstoffbriicken ist eine der ältesten und auch heute noch wichtigsten Anwendungsmöglichkeiten der I. Die am Beispiel der Assoziation der OH-Gruppe zu besprechenden Effekte lassen sich sinngemäß auf andere Protonendonatoren übertragen. Die OH-Valenzschwingung einer freien, nicht assoziierten OH-Gruppe ist im IR-Spektrum bei 3590 bis 3650 cm-1 zu finden. Durch Wasserstoffbrückenbindung wird eine Verschiebung nach kleineren Wellenzahlen bewirkt, wobei die OH-Bande gleichfalls breiter wird und an Intensität zunimmt. Die Verschiebung aus der ursprünglichen Lage ist um so größer, je stärker die gebildete H-Brücke ist. Da die Assoziation eine Gleichgewichtsreaktion ist, liegen in Abhängigkeit von Konzentration und Temperatur freie und assoziierte Alkohole nebeneinander vor. So erscheinen im Spektrum des Cyclohexanols in Tetrachlorkohlenstoff Banden bei 3615 bis 3620 cm-1 (nicht assoziierte OH-Gruppe), 3485 cm-1 (dimeres Assoziat) und 3320 cm-1 (höhere Assoziate) nebeneinander und in Abhängigkeit von der Konzentration mit unterschiedlicher Intensität (Abb. 9). Inter- und intramolekulare H-Brücken lassen sich leicht unterscheiden. Während erstere mit zunehmender Verdünnung der Lösung aufgebrochen werden, reagieren letztere darauf nicht.
5) Infrarot-Dichroismus. IR-Spektren, die mit polarisierter Strahlung erhalten werden, geben wertvolle Informationen über die Natur von Schwingungen und über die Orientierung von Gruppen innerhalb einer Probe. Bei der üblichen Anwendung der I. sind sowohl die Moleküle der Probe als auch der elektrische Vektor der Strahlung zufällig orientiert, so daß keine Polarisationseffekte beobachtet werden können. Untersuchungen an Einkristallen oder ausgerichteten Polymeren zeigen jedoch, daß die Bandenintensität stark von der Polarisationsrichtung der Strahlung abhängt. Dazu wird die Intensität A einer bestimmten Bande mit polarisiertem Licht, das einmal parallel, zum anderen senkrecht zur Orientierungsrichtung der Probe ausgerichtet ist, vermessen. Man erhält das dichroitische Verhältnis ![]()
das vom Grad der Orientierung der Probe sowie vom Winkel zwischen dem Übergangsmoment der Schwingung und der Orientierungsrichtung abhängig ist.
;)
Infrarotspektroskopie. Abb. 9: Konzentrationsabhängigkeit des IR-Spektrums von Cyclohexanol in Tetrachlorkohlenstoff.
6) Mittels der Matrix-Isolations-Technik können IR-Spektren von reaktionsfähigen Zwischenprodukten erhalten werden. Die zu untersuchenden Produkte werden in eine feste Matrix von Stickstoff, Kohlendioxid, Argon oder Xenon eingebettet, indem eine gasförmige Mischung, die eine kleine Menge der Probe, hauptsächlich aber die Matrixkomponente enthält, auf die Temperatur des flüssigen Wasserstoffs (20 K) oder des flüssigen Heliums (4 K) abgekühlt wird. So konnte z. B. das Radikal ClCO an seinen Absorptionsbanden bei 1880, 570 und 281 cm-1 nachgewiesen werden. Es wurde gebildet, indem Cl-Atome durch Photolyse von HCl in einer CO2-Matrix bei 14 K erzeugt wurden, die mit dem CO2 der Matrix zu obigem Radikal reagierten.
7) Spektroskopie im nahen IR. Von Wasserstoff enthaltenden Gruppen, wie N-H, C-H, O-H, werden im NIR zahlreiche Oberschwingungen und Kombinationsschwingungen beobachtet, die zur quantitativen Bestimmung solcher Gruppierungen (z. B. von Alkenen mit endständiger Doppelbindung oder cis-disubstituierten Doppelbindungen) sowie zur Untersuchung von Wasserstoffbrückenbindungen verwendet werden können.
8) Spektroskopie im fernen IR. Im FIR werden in direkter Fortsetzung der Anwendung im mittleren IR eine Reihe von Valenz- und Deformationsschwingungen von solchen Molekülen beobachtet, die aufgrund großer Atommassen im niederfrequenten Bereich absorbieren. Dies spielt z. B. bei der Untersuchung von Metallkomplexen eine wichtige Rolle, in denen Schwingungen zwischen Metall und Komplexbildner erfaßt werden können. Die in zahlreichen Systemen existierenden schwachen intermolekularen Wechselwirkungen rufen Schwingungen im FIR hervor. So finden sich die intermolekularen Schwingungen von H-Brücken bei 100 bis 200 cm-1, N-I-Valenzschwingungen in charge-transfer-Komplexen von Iod mit substituierten Pyridinen zwischen 65 und 95 cm-1.
9) Untersuchung von Adsorbenzien und adsorbierten Molekülen. Adsorptionsuntersuchungen werden mit konventionellen IR-Spektrometern unter Verwendung von Spezialküvetten durchgeführt. Meist mißt man von der entsprechend präparierten Probe das Absorptionsspektrum, seltener die Reflexionsspektren. Übliche Adsorbenzien sind Silicagel, Aluminiumoxid, Zeolithe, poröse Gläser u. a., die entweder allein oder mit feinverteilten Metallen oder Metalloxiden verwendet werden. Im Gegensatz zu anderen Methoden zur Oberflächenuntersuchung, die lediglich das durchschnittliche Verhalten des Systems wiedergeben, können mit Hilfe der IR-Spektren einzelne Spezies beobachtet werden. So findet man z. B. bei der Untersuchung poröser Gläser verschiedene OH-Schwingungen, von denen die Bande bei 3748 cm-1 einer freien Si-OH-Gruppe, die bei 3650 cm-1 einer assoziierten Si-OH-Gruppe und eine dritte Bande bei 3703 cm-1 einer an Bor gebundenen OH-Gruppe zugeordnet wird. Auch die Spektren der adsorbierten Moleküle erlauben Aussagen über die Oberfläche von Adsorbenzien, z. B. wird NH3 an einem Silicium-Aluminium-Katalysator als physikalisch gebundenes NH3 sowie als koordinativ gebundenes NH3 und als NH4+ adsorbiert. Aus der relativen Intensität der entsprechenden Banden ließ sich das Verhältnis von Lewis- und Brönsted-Zentren auf der Katalysatoroberfläche zu 4 : 1 bestimmen.
Wesentliche Beiträge konnten IR-Untersuchungen zum Mechanismus der Hydrierung von Olefinen an Nickelkatalysatoren leisten. So zeigte am Katalysator adsorbiertes Ethen Banden bei 2800 und 2890 cm-1, die einer Ni-CH2-CH2-Ni-Gruppe zugeordnet wurden.
;)
Infrarotspektroskopie. Abb. 10: Methoden der abgeschwächten Totalreflexion: (a) Einfachreflexion, (b) Mehrfachreflexion.
10) Die Methode der abgeschwächten Totalreflexion (ATR-Technik, Abk. von attenuated total reflectance) dient zur Untersuchung solcher Proben, die aufgrund ihrer begrenzten Durchlässigkeit für IR-Strahlung mit der üblichen Technik nicht vermessen werden können, z. B. zur Untersuchung von Beschichtungen auf den verschiedenartigsten Materialien sowie von Festkörpern, wie Folien, Fasern, Kunststoffen, Leder und Gummi. Als Theorie liegt dieser Methode zugrunde, daß die auf die Hypotenusenfläche eines Prismas fallende Strahlung dort totalreflektiert wird. Dabei dringt sie ein wenig in die angrenzende Probe ein. Wenn diese Probe eine Substanz enthält, die IR-Strahlung absorbiert, so wird die totalreflektierte Strahlung bei diesen Wellenlängen eine Schwächung erfahren. Trägt man die Intensität der reflektierten Strahlung als Funktion der Wellenlänge auf, so erhält man ein Spektrum, das weitgehend dem Absorptionsspektrum der Substanz entspricht. Durch Mehrfachreflexion kann der Effekt verstärkt werden (Abb. 10a und b).







Wenn Sie inhaltliche Anmerkungen zu diesem Artikel haben, können Sie die Redaktion per E-Mail informieren. Wir lesen Ihre Zuschrift, bitten jedoch um Verständnis, dass wir nicht jede beantworten können.